Coverage in gene regions
Briana Mittleman
2017-11-20
Last updated: 2017-11-30
Code version: 35b1aa1
Initial exploration for data format
The goal of this analysis is too look at read coverage in genes/exons/and in genes + promoter regions for my Net-seq data and the Mayer data. This will help me understand if the problem with this data is coverage.
I have downloaded 3 bed files from the UCSC table browser:
Ref seq genes
Ref seq genes + 250 upstream
Ref seq exons
I will use the bedtools coverage function. Documentation is below:
http://bedtools.readthedocs.io/en/latest/content/tools/coverage.html
- a bed file for the genes/exons
- bam files for my data and the mayer data
bedtools coverage -a <FILE> \
-b <FILE1, FILE2, ..., FILEN>Write a bash script for genes overlap: genome_overlap.sh
#!/bin/bash
#SBATCH --job-name=bedtools_coverage
#SBATCH --time=8:00:00
#SBATCH --partition=broadwl
#SBATCH --mem=8G
#SBATCH --tasks-per-node=4
#$1 reference to use
bedtools coverage -a $1 -b /project2/gilad/briana/Net-seq/Net-seq1/data/sort/YG-SP-NET1-18486-dep-2017-10-13_S4_R1_001-sort.bam /project2/gilad/briana/Net-seq/Net-seq1/data/sort/YG-SP-NET1-18508-dep-2017-10-13_S2_R1_001-sort.bam /project2/gilad/briana/Net-seq/Net-seq1/data/sort/YG-SP-NET1-18508-nondep-2017-10-13_S3_R1_001-sort.bam /project2/gilad/briana/Net-seq/Net-seq1/data/sort/YG-SP-NET1-Unk1_S6_R1_001-sort.bam /project2/gilad/briana/Net-seq/data/sort/SRR1575922-sort.bam
#> to a txt file
bedtools coverage -a ref_seq_gene_hg19_ -b /project2/gilad/briana/Net-seq/Net-seq1/data/net1_18486_dep_dedup_chr.bed -hist > gene_coverage_18486_dedup_hist.txt
Try with only 1 file first.
Try with /project2/gilad/briana/Net-seq/Net-seq1/data/net1_18486_dep_dedup_chr.bed
This works but it is for the deduplicated file. I am going to do the sorted files first. I need to convert the sorted bam files to bed files. All of the sorted bed files with the chr label are in data/bed
Should probably do this for the deduplicated files as well.
bedtools bamtobed [OPTIONS] -i <BAM>
awk '$0="chr"$0' file > new_file
bedtools bamtobed -i file.bam | awk '$0="chr"$0' > newfile.bedOnly want first 6 columns of the ref file
cat ref_seq_gene_hg19 | cut -f 1-6 > ref_seq_gene_hg19_small_cut
Still to big. Run on one file.
bedtools coverage -a ref_seq_gene_hg19_small_cut -b /project2/gilad/briana/Net-seq/Net-seq1/data/bed/net1_18486_dep_chr.bed > gene_coverage_18486_hist.txtRun this to make:
gene_coverage_18508_dep_hist.txt
gene_coverage_18508_dep_hist.txt
gene_coverage_18508_nondep_hist.txt
gene_coverage_19238_dep_hist.txt
gene_coverage_mayer_SRR1575922_hist.txt
Sort the bed files then rerun the coverage with the counts option to get one read count per gene. I have the script to sort the script files in each data/bed folder. ls
bedtools coverage -counts -a ref_seq_gene_hg19_small_cut -b /project2/gilad/briana/Net-seq/Net-seq1/data/bed_sort/net1_18486_dep_chr_sort.bed > gene_cov_count/gene_coverage_18486_count.txt
bedtools coverage -counts -a ref_seq_gene_hg19_small_cut -b /project2/gilad/briana/Net-seq/Net-seq1/data/bed_sort/net1_18508_dep_chr_sort.bed > gene_cov_count/gene_coverage_18508_dep_count.txt
bedtools coverage -counts -a ref_seq_gene_hg19_small_cut -b /project2/gilad/briana/Net-seq/Net-seq1/data/bed_sort/net1_18508_nondep_chr_sort.bed > gene_cov_count/gene_coverage_18508_nondep_count.txt
bedtools coverage -counts -a ref_seq_gene_hg19_small_cut -b /project2/gilad/briana/Net-seq/Net-seq1/data/bed_sort/net1_19238_dep_chr_sort.bed > gene_cov_count/gene_coverage_19238_dep_count.txt
bedtools coverage -counts -a ref_seq_gene_hg19_small_cut -b /project2/gilad/briana/Net-seq/data/bed_sort/mayer_SRR1575922_chr_sort.bed > gene_cov_count/gene_coverage_mayer_SRR1575922_count.txt
Data input (non-depuplicated)
Gene Coverage
Install packages:
library(vioplot)Loading required package: smPackage 'sm', version 2.2-5.4: type help(sm) for summary informationlibrary(dplyr)
Attaching package: 'dplyr'The following objects are masked from 'package:stats':
filter, lagThe following objects are masked from 'package:base':
intersect, setdiff, setequal, unionlibrary(ggplot2)
library(lattice)gene_coverage_18486_count= read.csv("../data/gene_cov_count/gene_coverage_18486_count.txt", header=FALSE, sep="\t")
gene_coverage_18508_dep_count= read.csv("../data/gene_cov_count/gene_coverage_18508_dep_count.txt", header=FALSE, sep="\t")
gene_coverage_18508_nondep_count= read.csv("../data/gene_cov_count/gene_coverage_18508_nondep_count.txt", header=FALSE, sep="\t")
gene_coverage_19238_dep_count= read.csv("../data/gene_cov_count/gene_coverage_19238_dep_count.txt", header=FALSE, sep="\t")
gene_coverage_mayer_dep_count = read.csv("../data/gene_cov_count/gene_coverage_mayer_SRR1575922_count.txt", header=FALSE, sep="\t")colnames(gene_coverage_18486_count) = c("chr", "start", "end", "name", "score", "strand", "counts")
colnames(gene_coverage_18508_dep_count)= c("chr", "start", "end", "name", "score", "strand", "counts")
colnames(gene_coverage_18508_nondep_count)=c("chr", "start", "end", "name", "score", "strand", "counts")
colnames(gene_coverage_19238_dep_count)= c("chr", "start", "end", "name", "score", "strand", "counts")
colnames(gene_coverage_mayer_dep_count)= c("chr", "start", "end", "name", "score", "strand", "counts")Next I will merge theses files to create 1 file with all of the data.
gene_coverage_all= cbind(gene_coverage_18486_count, gene_coverage_18508_dep_count$counts, gene_coverage_18508_nondep_count$counts, gene_coverage_19238_dep_count$counts, gene_coverage_mayer_dep_count$counts)
colnames(gene_coverage_all)= c("chr", "start", "end", "name", "score", "strand", "c_18486", "c_18508_dep", "c_18508_nondep", "c_19238_dep", "c_mayer")Add a column for length of A:
#add length column
gene_coverage_all= mutate(gene_coverage_all, length=end-start)
#add columns for standard count
gene_coverage_all= mutate(gene_coverage_all,st_18486=c_18486/length)
gene_coverage_all= mutate(gene_coverage_all,st_18508_dep= c_18508_dep /length)
gene_coverage_all= mutate(gene_coverage_all,st_18508_nondep=c_18508_nondep/length)
gene_coverage_all= mutate(gene_coverage_all,st_19238=c_19238_dep/length)
gene_coverage_all= mutate(gene_coverage_all,st_mayer=c_mayer/length)plot(sort(log(gene_coverage_all$st_18486 + .1), decreasing=TRUE), ylab="log of gene count standardized by length", xlab="Genes", main="18486")
abline(a=log(mean(gene_coverage_all$st_18486 + .1)),b=0)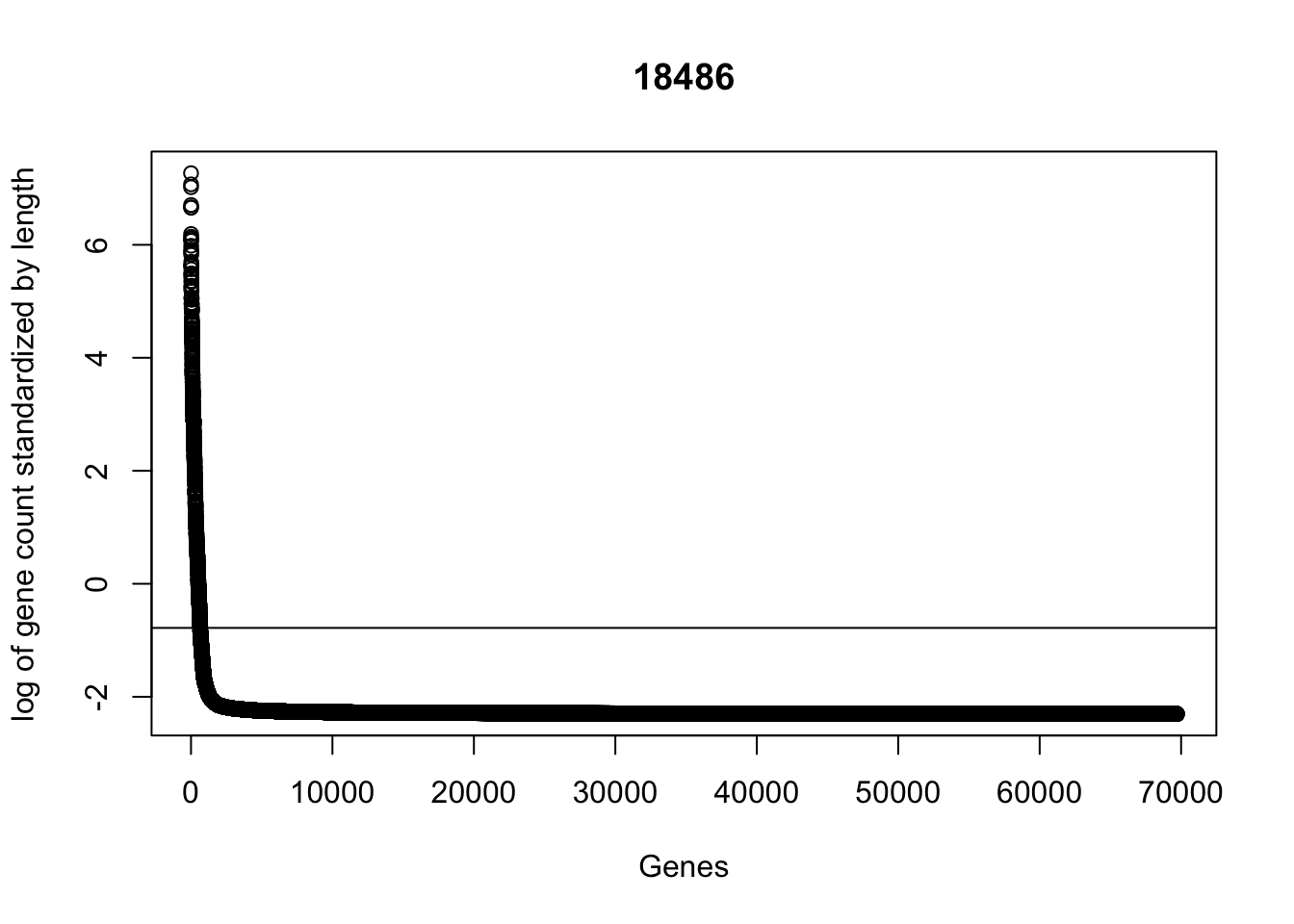
plot(sort(log(gene_coverage_all$st_18508_dep + .1), decreasing=TRUE), ylab="log of gene count standardized by length", xlab="Genes", main="18505 dep")
abline(a=log(mean(gene_coverage_all$st_18508_dep + .1)),b=0)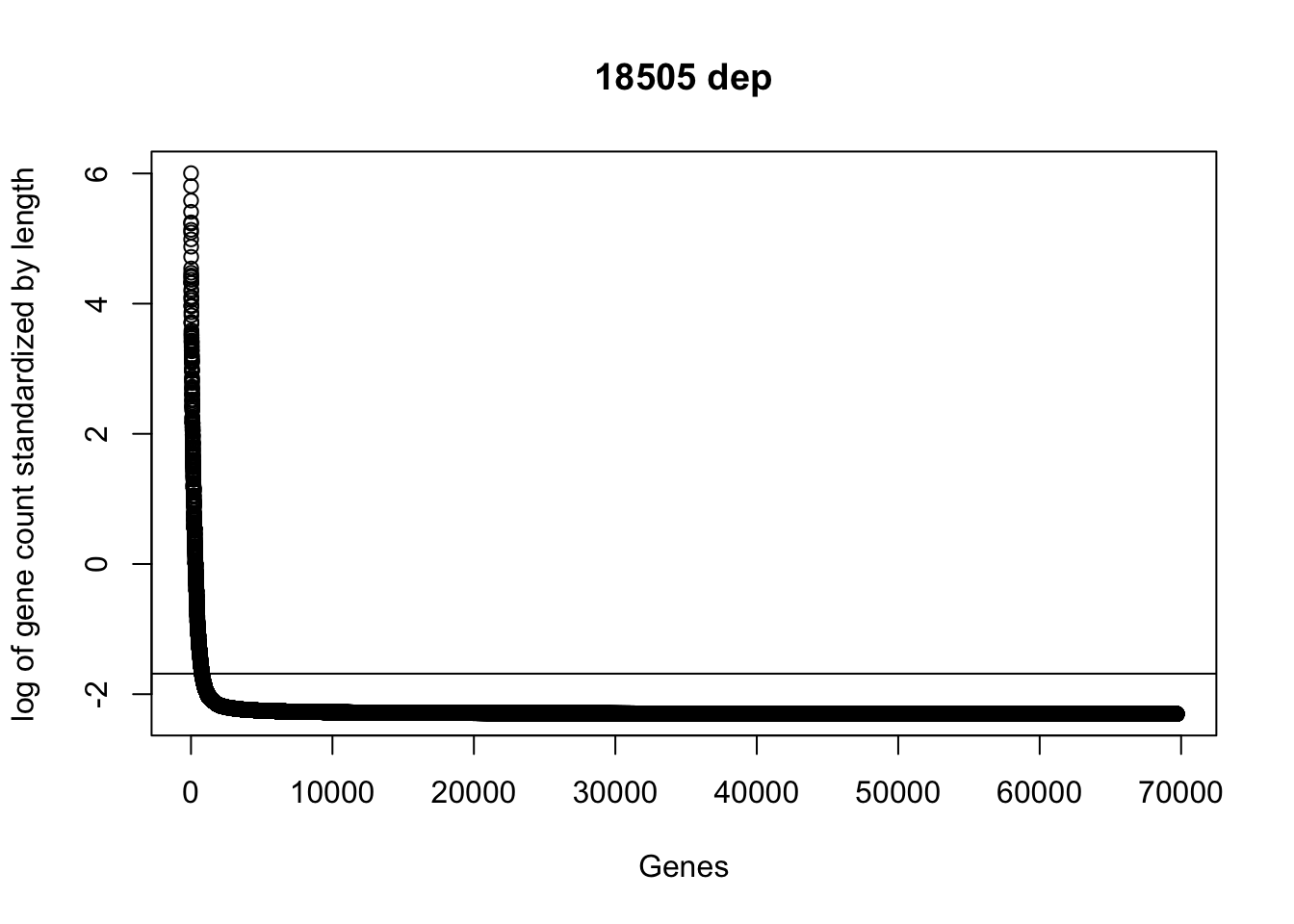
plot(sort(log(gene_coverage_all$st_18508_nondep + .1), decreasing=TRUE), ylab="log of gene count standardized by length", xlab="Genes", main="18505 nondep")
abline(a=log(mean(gene_coverage_all$st_18508_nondep +.1)),b=0)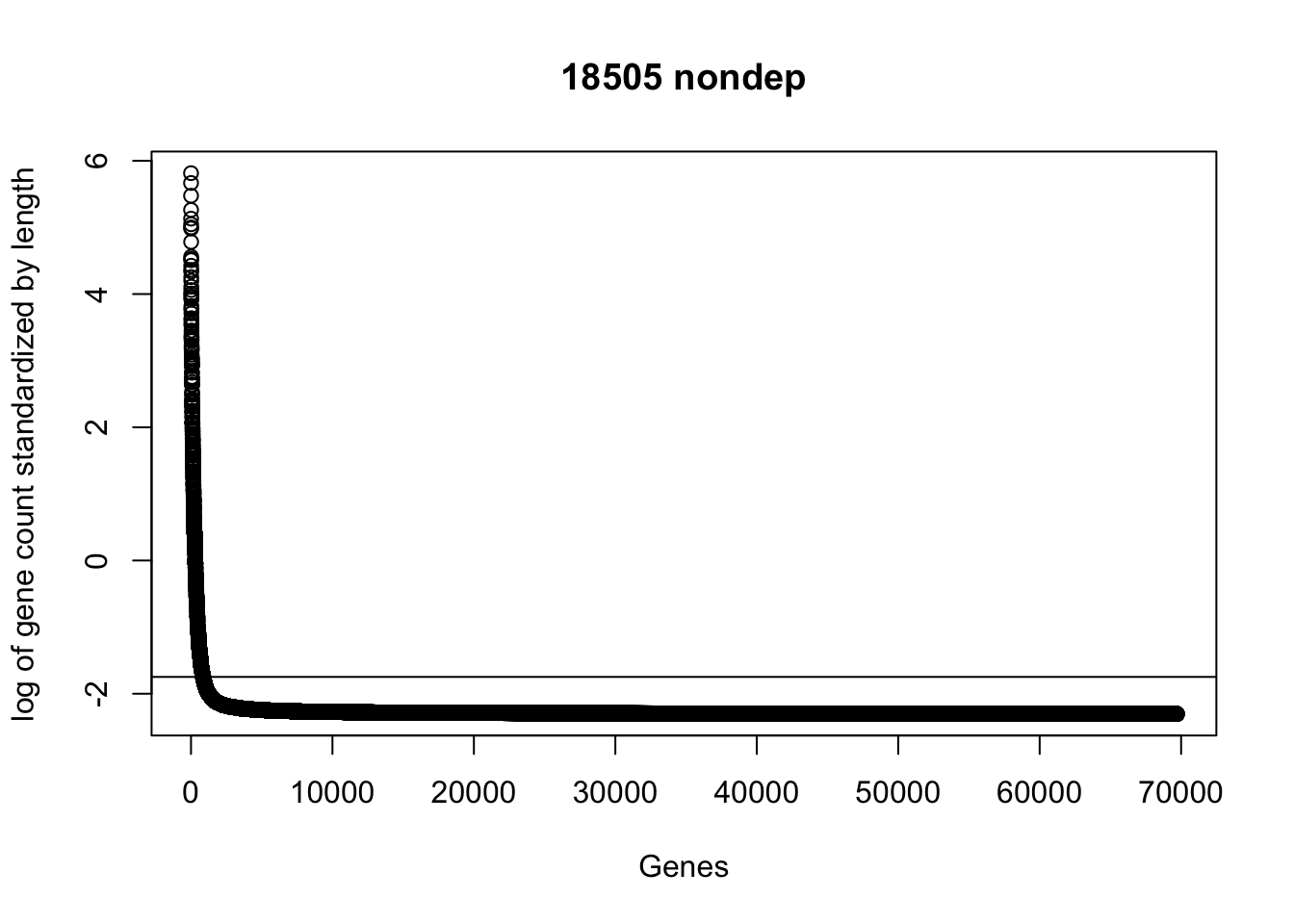
plot(sort(log(gene_coverage_all$st_19238 + .1), decreasing=TRUE), ylab="log of gene count standardized by length", xlab="Genes", main="19238")
abline(a=log(mean(gene_coverage_all$st_19238 + .1)),b=0)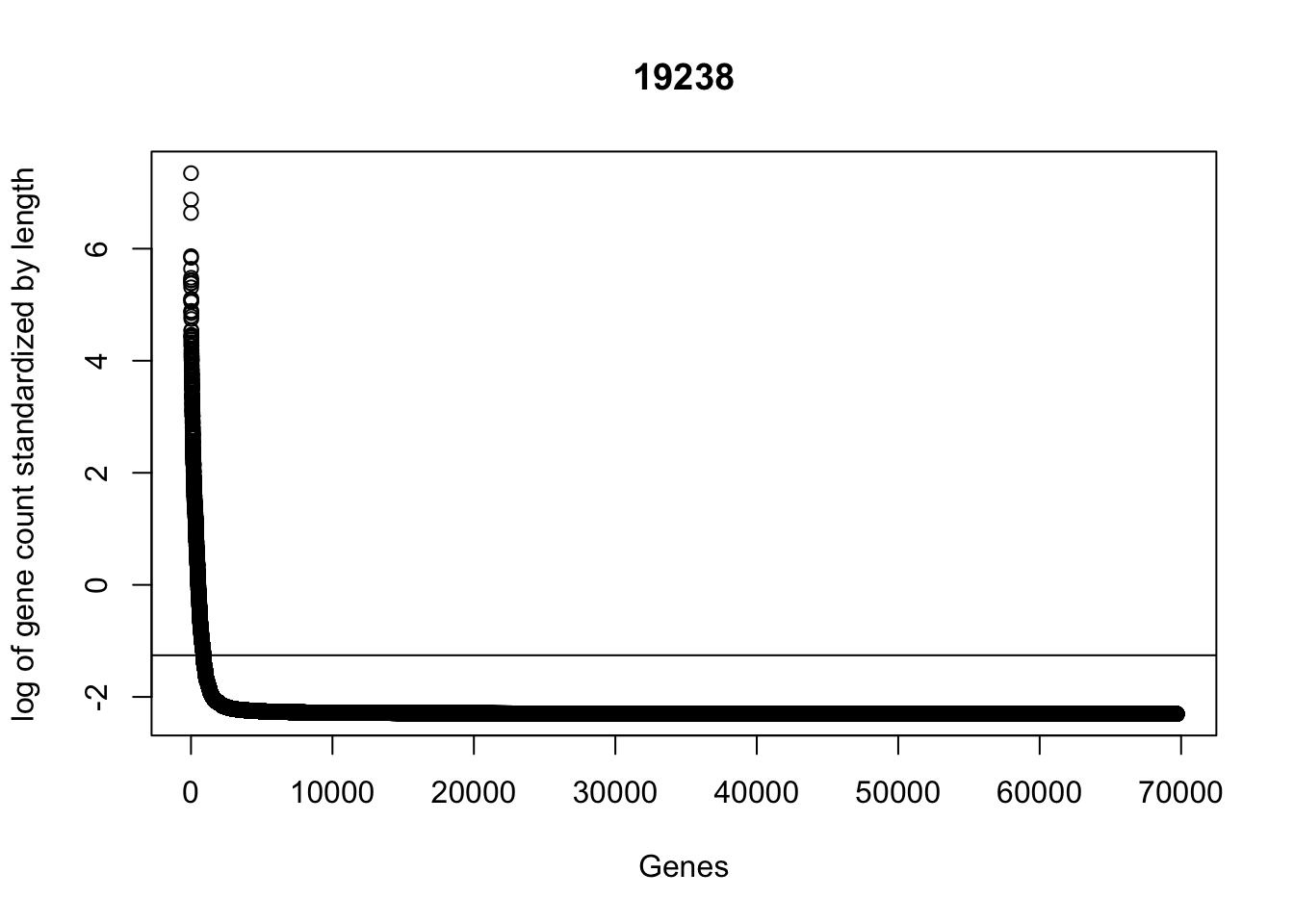
plot(sort(log(gene_coverage_all$st_mayer + .1), decreasing=TRUE), ylab="log of gene count standardized by length", xlab="Genes", main="Mayer")
abline(a=log(mean(gene_coverage_all$st_mayer + .1)),b=0)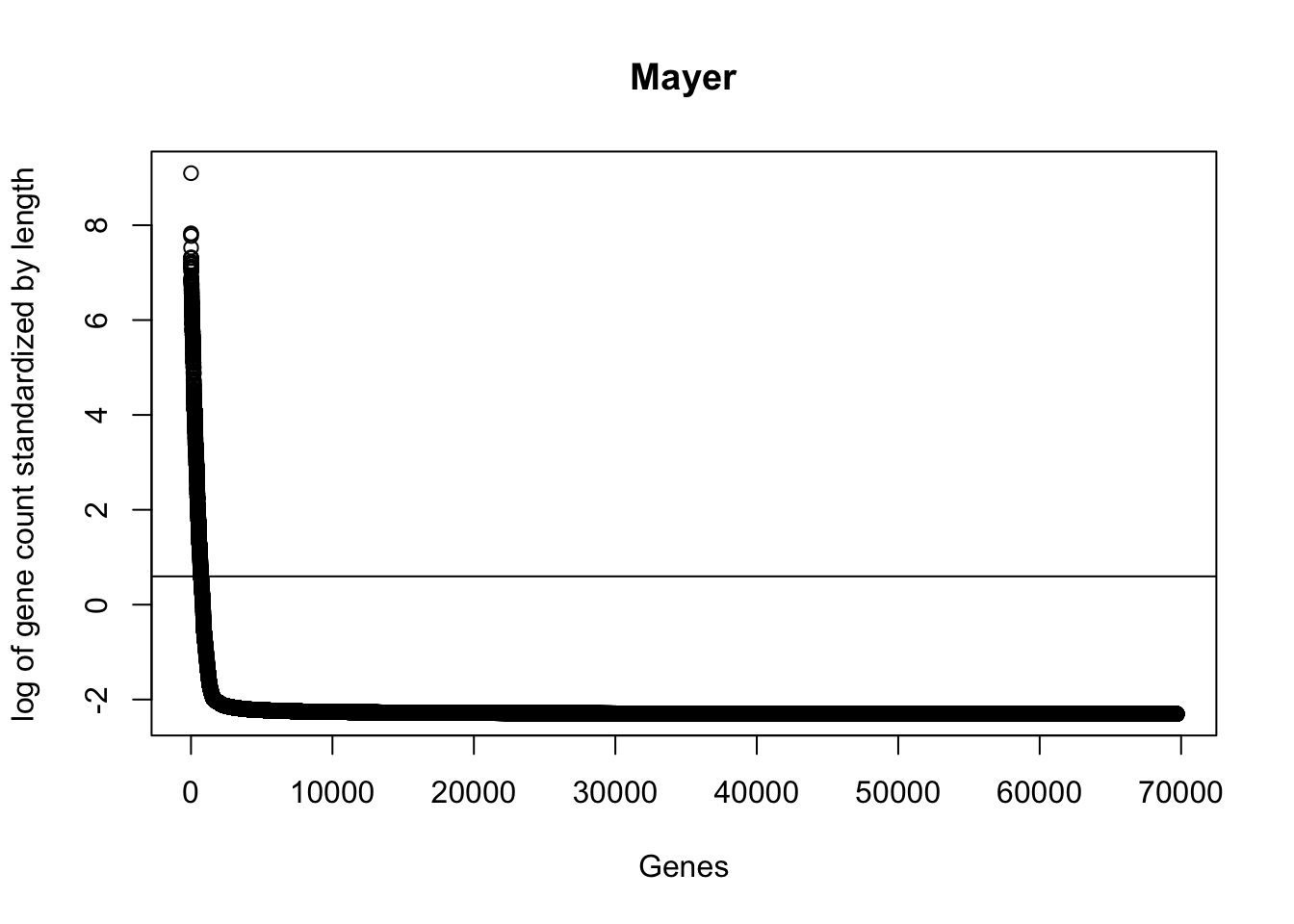
#boxplot- violin plot is better but you get infinities
boxplot(-log(gene_coverage_all$st_18486), -log(gene_coverage_all$st_18508_dep), -log(gene_coverage_all$st_18508_nondep), -log(gene_coverage_all$st_19238), -log(gene_coverage_all$st_mayer), names=c("18486", "18508_dep", "18508_nondep", "19238", "Mayer"), ylab="-log of standard expression")Warning in bplt(at[i], wid = width[i], stats = z$stats[, i], out = z$out[z
$group == : Outlier (Inf) in boxplot 1 is not drawnWarning in bplt(at[i], wid = width[i], stats = z$stats[, i], out = z$out[z
$group == : Outlier (Inf) in boxplot 2 is not drawnWarning in bplt(at[i], wid = width[i], stats = z$stats[, i], out = z$out[z
$group == : Outlier (Inf) in boxplot 3 is not drawnWarning in bplt(at[i], wid = width[i], stats = z$stats[, i], out = z$out[z
$group == : Outlier (Inf) in boxplot 4 is not drawnWarning in bplt(at[i], wid = width[i], stats = z$stats[, i], out = z$out[z
$group == : Outlier (Inf) in boxplot 5 is not drawn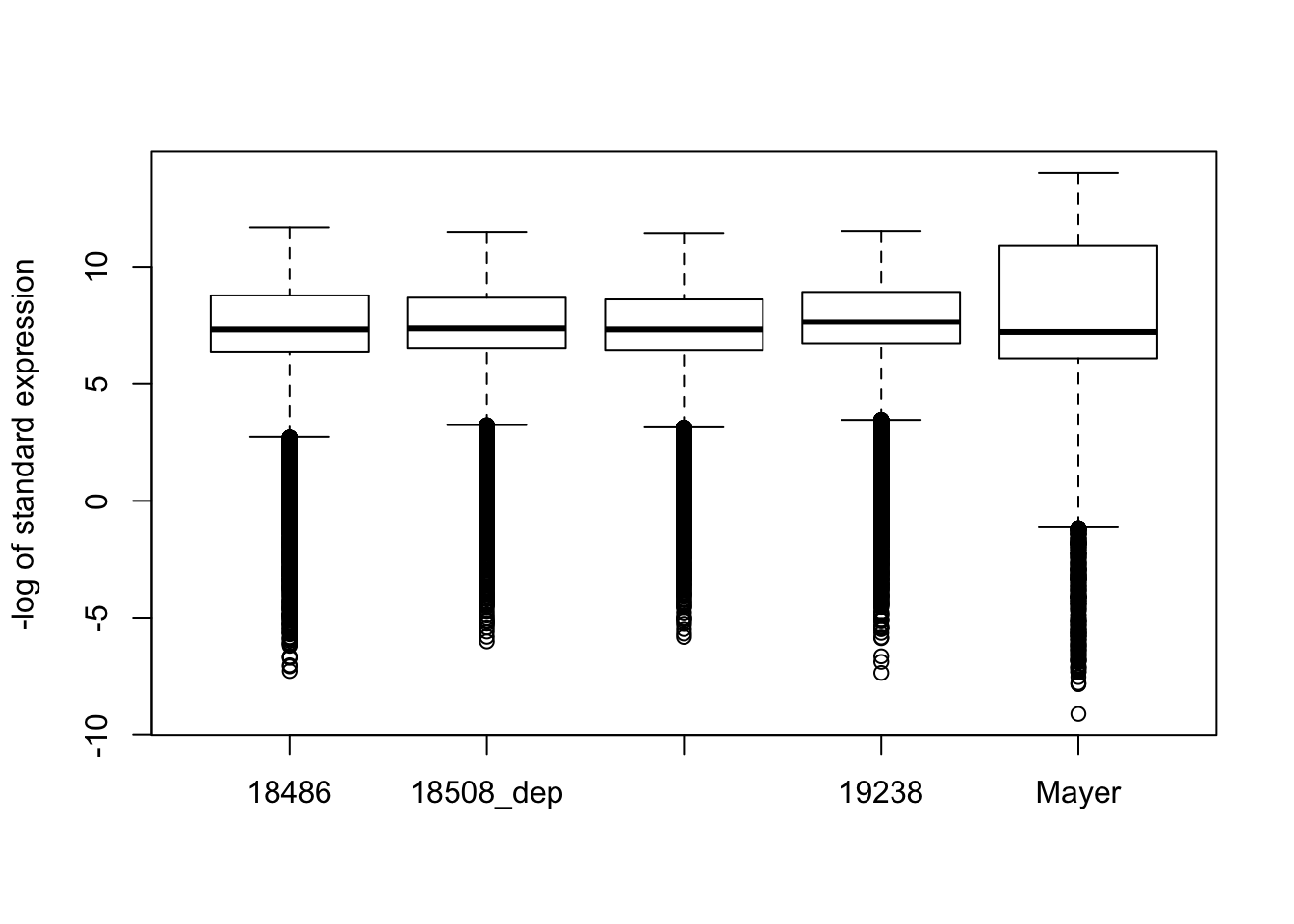
Scatter plots:
par(mfrow = c(1,2))
plot(gene_coverage_all$st_18486 ~ gene_coverage_all$st_18508_dep, xlab="18486", ylab = "18508")
abline(lm(gene_coverage_all$st_18486 ~ gene_coverage_all$st_18508_dep))
plot(gene_coverage_all$st_18486 ~ gene_coverage_all$st_mayer, xlab= "18486", ylab="Mayer")
abline(lm(gene_coverage_all$st_18486 ~ gene_coverage_all$st_mayer))
title("Standard Count Correlatation") 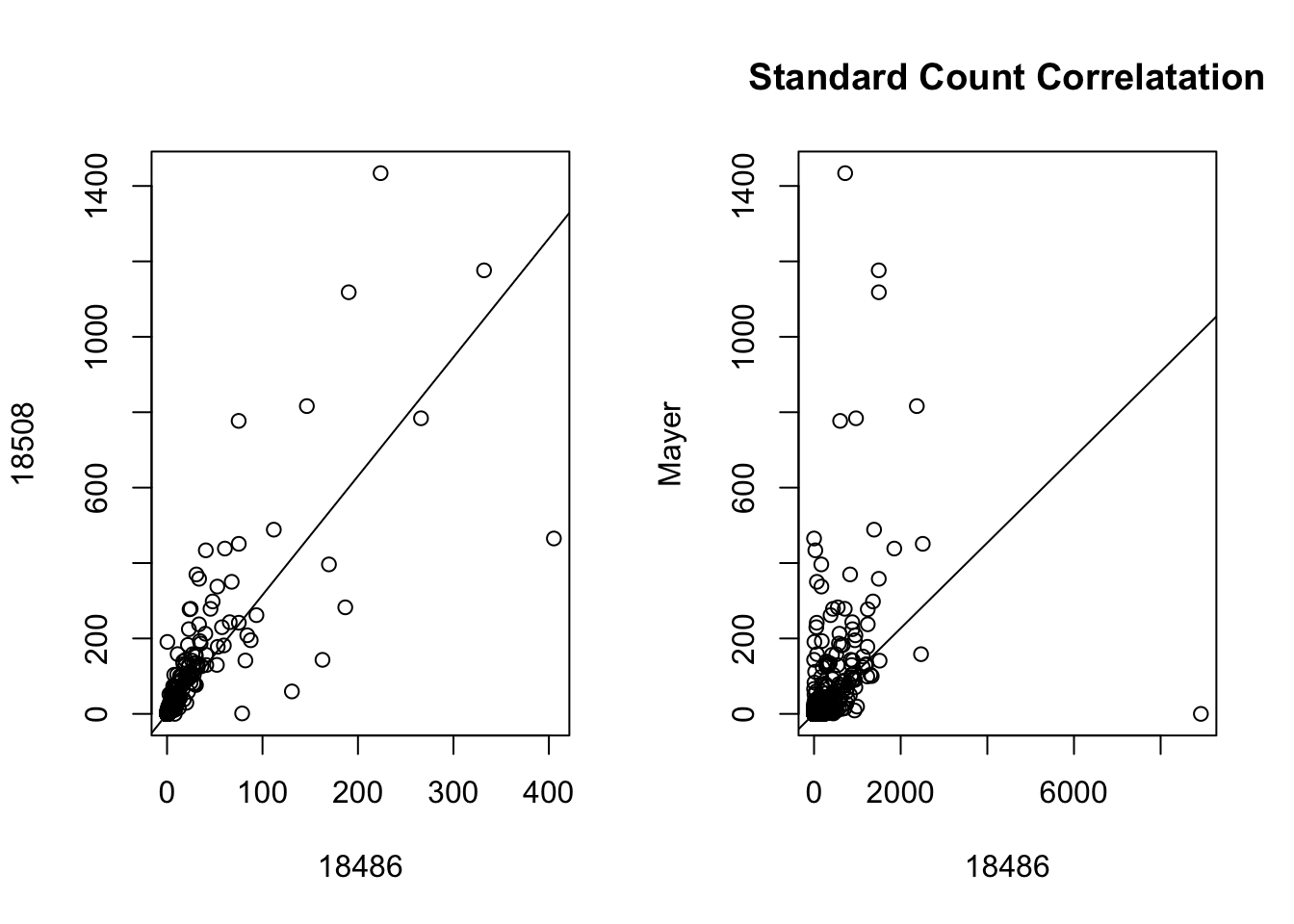
To use ggplot I need to reformat the dataframe to gene by sample.
gene_names= gene_coverage_all$name
standard_counts= gene_coverage_all[, 13:17]
standard_counts=cbind(gene_names, standard_counts)
#standard_counts_t=as.data.frame(standard_counts %>% t)ggplot(standard_counts, aes(x=log(st_18486 + .1))) + geom_density()
ggplot(standard_counts, aes(x=log(st_18508_dep + .1))) + geom_density()
ggplot(standard_counts, aes(x=log(st_18508_nondep + .1))) + geom_density()
ggplot(standard_counts, aes(x=log(st_19238 +.1))) + geom_density()
ggplot(standard_counts, aes(x=log(st_mayer + .1))) + geom_density()
Next step: subset the overexpressed genes to see distribution. First I just cut the top 5000.
This is probably the snoRNA and rRNAs.
Next: NR ncRNA
coding_genes= standard_counts[ grepl("NR", standard_counts$gene_names)==FALSE,]
ggplot(coding_genes, aes(x=log(st_18486 + .1))) + geom_density()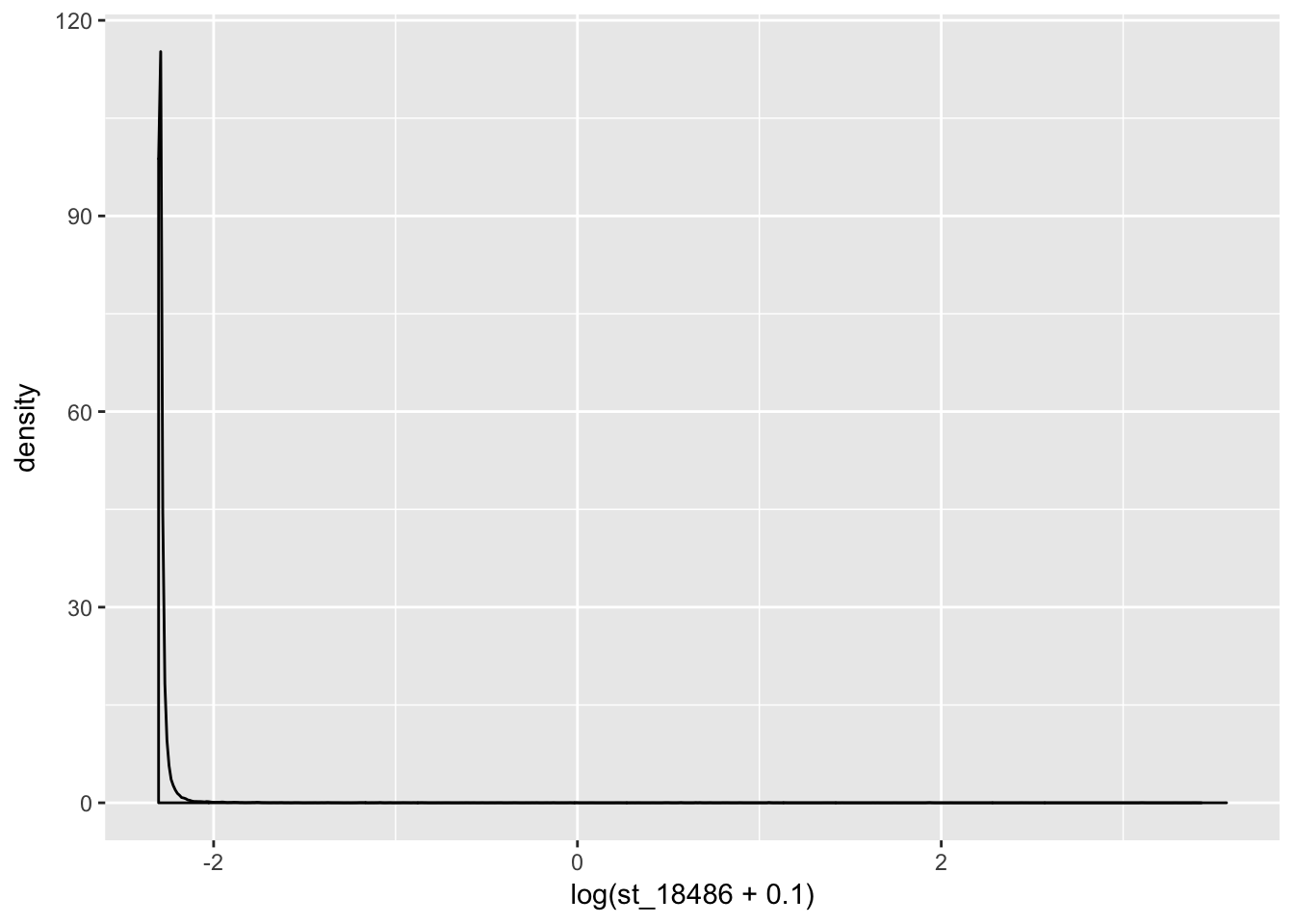
ggplot(coding_genes, aes(x=log(st_18508_dep + .1))) + geom_density()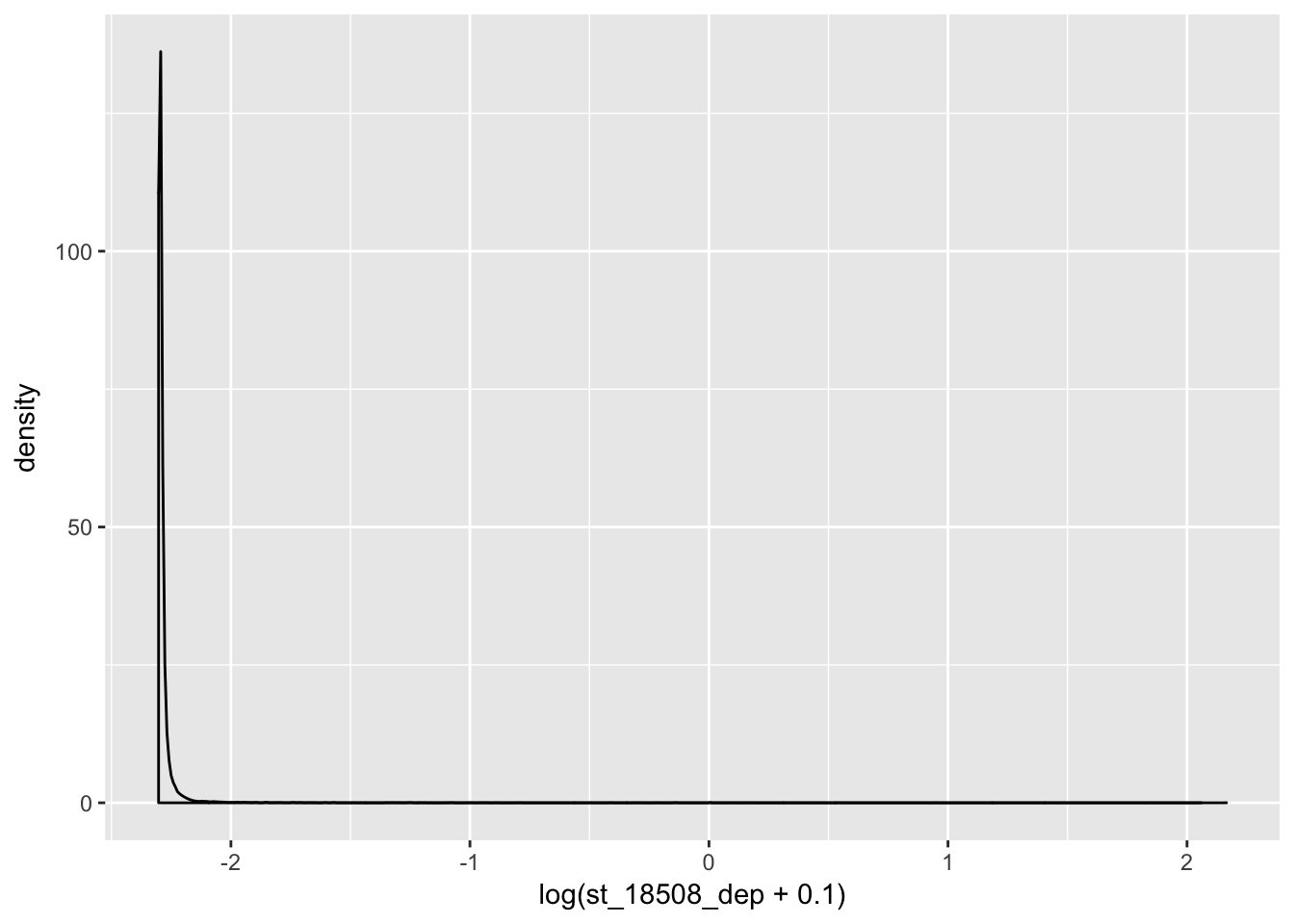
ggplot(coding_genes, aes(x=log(st_18508_nondep + .1))) + geom_density()
ggplot(coding_genes, aes(x=log(st_19238 +.1))) + geom_density()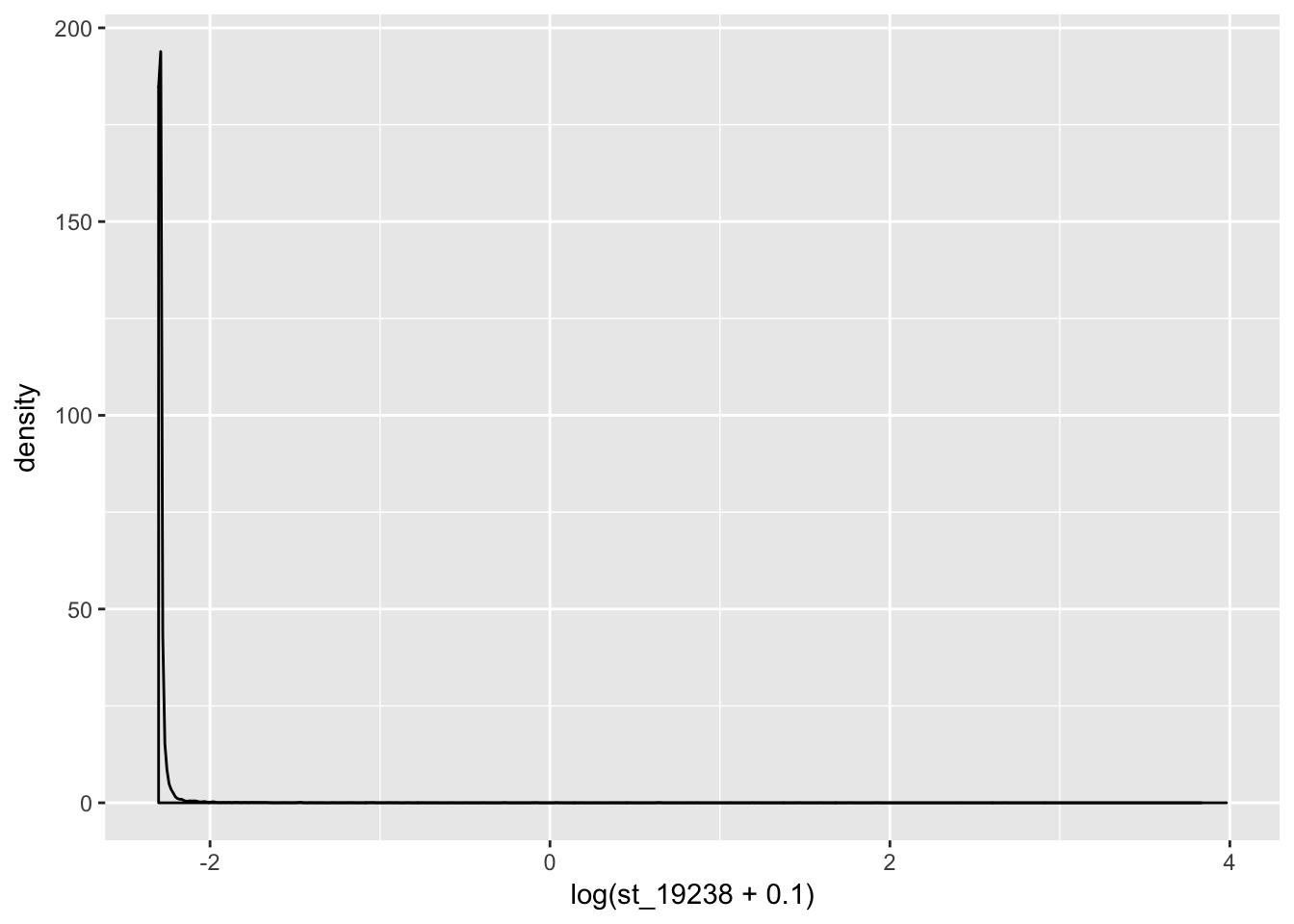
ggplot(coding_genes, aes(x=log(st_mayer + .1))) + geom_density()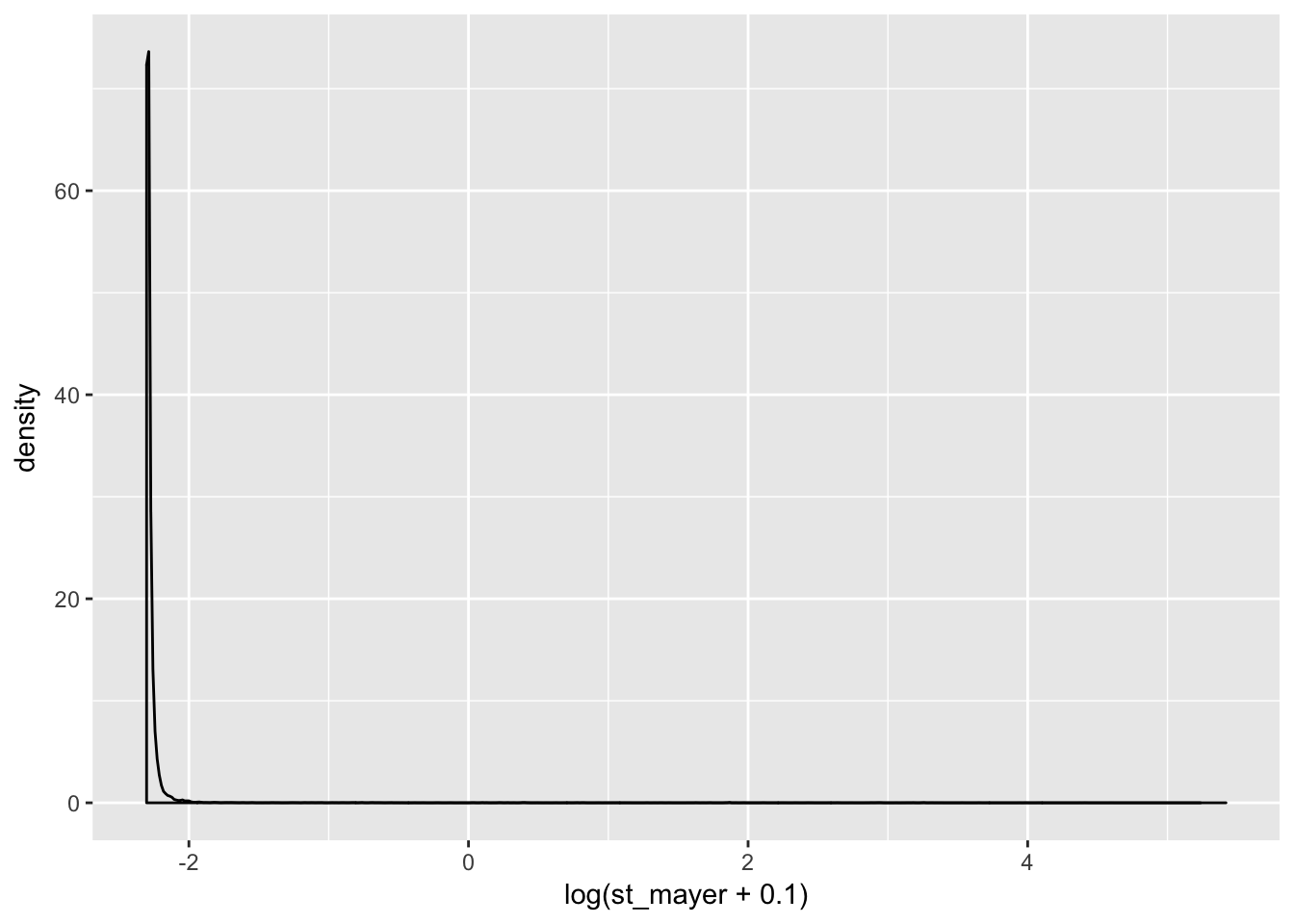
Remove zeros:
#ggplot(coding_genes, aes(x= log(st_18486[coding_genes$st_18486 > 0]))) + geom_density()sorted_18486=sort(log(standard_counts$st_18486 + .1), decreasing=TRUE)
sorted_18508_dep=sort(log(standard_counts$st_18508_dep + .1), decreasing=TRUE)
sorted_18508_nondep=sort(log(standard_counts$st_18508_nondep + .1), decreasing=TRUE)
sorted_19238=sort(log(standard_counts$st_19238 + .1), decreasing=TRUE)
sorted_mayer=sort(log(standard_counts$st_mayer + .1), decreasing=TRUE)
sorted_18486_cut= sorted_18486[5000:length(sorted_18486)]
sorted_18508_dep_cut= sorted_18508_dep[5000:length(sorted_18508_dep)]
sorted_184508_nondep_cut= sorted_18508_nondep[5000:length(sorted_18508_nondep)]
sorted_19238_cut= sorted_19238[5000:length(sorted_19238)]
sorted_mayer_cut= sorted_mayer[5000:length(sorted_mayer)]
dat=cbind.data.frame(sorted_18486_cut,sorted_18508_dep_cut, sorted_184508_nondep_cut, sorted_19238_cut,sorted_mayer_cut )
ggplot(dat, aes(x=sorted_18486_cut)) + geom_density()
ggplot(dat, aes(x=sorted_18508_dep_cut)) + geom_density()
ggplot(dat, aes(x=sorted_184508_nondep_cut)) + geom_density()
ggplot(dat, aes(x=sorted_19238_cut)) + geom_density()
ggplot(dat, aes(x=sorted_mayer_cut)) + geom_density()
Zeros are represented as -2.3
Propotion of detected genes
Take the sum of the genes that do not equal zero for each set.
prop_gene_non0_18486= sum(gene_coverage_all$c_18486 !=0) /length(gene_coverage_all$c_18486)
prop_gene_non0_18508dep= sum(gene_coverage_all$c_18508_dep !=0) /length(gene_coverage_all$c_18508_dep)
prop_gene_non0_18508_nondep= sum(gene_coverage_all$c_18508_nondep !=0) /length(gene_coverage_all$c_18508_nondep)
prop_gene_non0_19238= sum(gene_coverage_all$c_19238_dep !=0) /length(gene_coverage_all$c_19238_dep)
prop_gene_non0_mayer= sum(gene_coverage_all$c_mayer !=0) /length(gene_coverage_all$c_mayer)
prop_non0_all= c(prop_gene_non0_18486,prop_gene_non0_18508dep, prop_gene_non0_18508_nondep, prop_gene_non0_19238, prop_gene_non0_mayer)
barplot(prop_non0_all, names=c("18486", "18505 \n dep", "18505 \n non dep", "19238", "Mayer"), ylab= "Proportion of detected genes", ylim=0:1, xlab= "Library")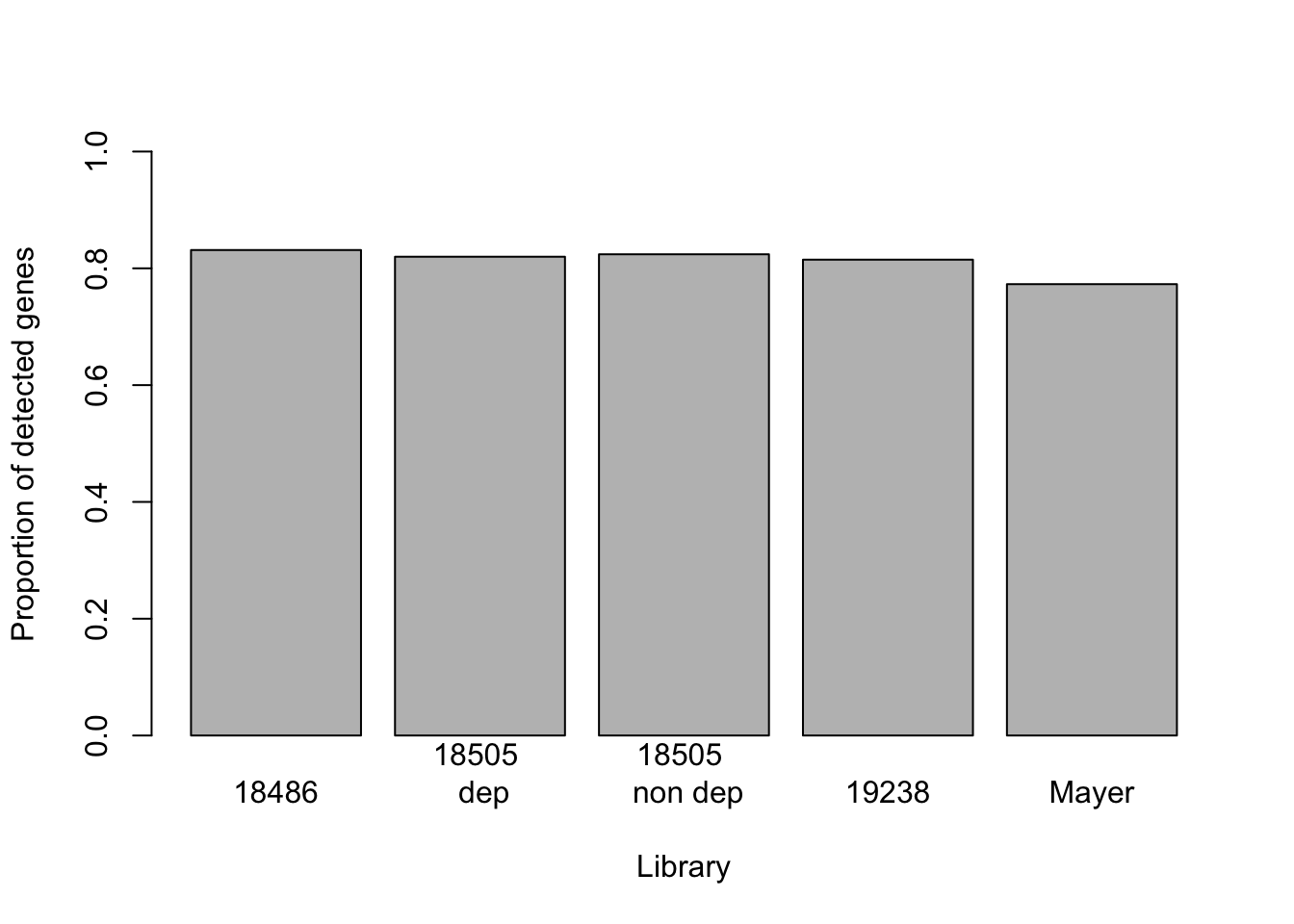
Gene and promoter coverage
prom_coverage_18486_count= read.csv("../data/prom_coverage/prom_coverage_18486_count.txt", header=FALSE, sep="\t")
prom_coverage_18508_dep_count= read.csv("../data/prom_coverage/prom_coverage_18508_dep_count.txt", header=FALSE, sep="\t")
prom_coverage_18508_nondep_count= read.csv("../data/prom_coverage/prom_coverage_18508_nondep_count.txt", header=FALSE, sep="\t")
prom_coverage_19238_dep_count= read.csv("../data/prom_coverage/prom_coverage_19238_dep_count.txt", header=FALSE, sep="\t")
prom_coverage_mayer_dep_count = read.csv("../data/prom_coverage/promoter_coverage_mayer_SRR1575922_count.txt", header=FALSE, sep="\t")colnames(prom_coverage_mayer_dep_count)= c("chr", "start", "end", "name", "score", "strand", "counts")
colnames(prom_coverage_19238_dep_count)= c("chr", "start", "end", "name", "score", "strand", "counts")
colnames(prom_coverage_18508_nondep_count)= c("chr", "start", "end", "name", "score", "strand", "counts")
colnames(prom_coverage_18508_dep_count) = c("chr", "start", "end", "name", "score", "strand", "counts")
colnames(prom_coverage_18486_count)= c("chr", "start", "end", "name", "score", "strand", "counts")Next I will merge theses files to create 1 file with all of the data.
prom_coverage_all= cbind(prom_coverage_18486_count, prom_coverage_18508_dep_count$counts, prom_coverage_18508_nondep_count$counts, prom_coverage_19238_dep_count$counts, prom_coverage_mayer_dep_count$counts)
colnames(prom_coverage_all)= c("chr", "start", "end", "name", "score", "strand", "c_18486", "c_18508_dep", "c_18508_nondep", "c_19238_dep", "c_mayer")Add a column for length of A and standardize the counts. NOTE would it be better to standardize by the gene and the promoter or just the gene length?
#add length column
prom_coverage_all= mutate(prom_coverage_all, length=end-start)
#add columns for standard count
prom_coverage_all= mutate(prom_coverage_all,st_18486=c_18486/length)
prom_coverage_all= mutate(prom_coverage_all,st_18508_dep= c_18508_dep /length)
prom_coverage_all= mutate(prom_coverage_all,st_18508_nondep=c_18508_nondep/length)
prom_coverage_all= mutate(prom_coverage_all,st_19238=c_19238_dep/length)
prom_coverage_all= mutate(prom_coverage_all,st_mayer=c_mayer/length)par(mfrow = c(3,2))
plot(sort(log(prom_coverage_all$st_18486 + .1), decreasing=TRUE), ylab="-log of gene count standardized by length", xlab="Genes + Prom", main="18486")
abline(a=log(mean(prom_coverage_all$st_18486 + .1)),b=0)
plot(sort(log(prom_coverage_all$st_18508_dep + .1), decreasing=TRUE), ylab="-log of gene count standardized by length", xlab="Genes + Prom", main="18505 dep")
abline(a=log(mean(prom_coverage_all$st_18508_dep + .1)),b=0)
plot(sort(log(prom_coverage_all$st_18508_nondep + .1), decreasing=TRUE), ylab="-log of gene count standardized by length", xlab="Genes + prom", main="18505 nondep")
abline(a=log(mean(prom_coverage_all$st_18508_nondep + .1)),b=0)
plot(sort(log(prom_coverage_all$st_19238 + .1) , decreasing=TRUE), ylab="-log of gene count standardized by length", xlab="Genes + prom", main="19238")
abline(a=log(mean(prom_coverage_all$st_19238 + .1)),b=0)
plot(sort(log(prom_coverage_all$st_mayer + .1), decreasing=TRUE), ylab="-log of gene count standardized by length", xlab="Genes + prom", main="Mayer")
abline(a=log(mean(prom_coverage_all$st_mayer + .1)),b=0)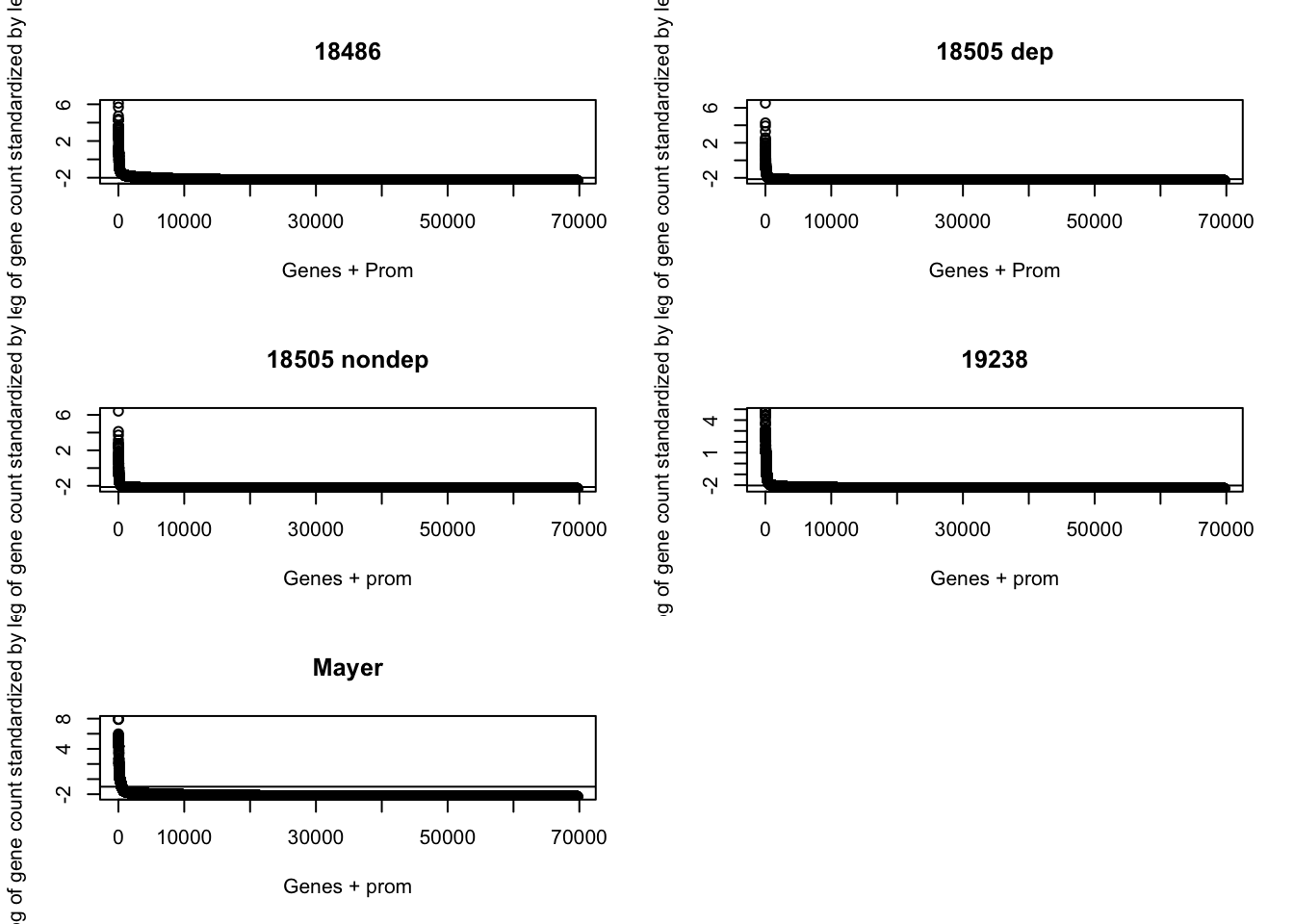
I beleive this shows that the promoters do not hold a majority of the reads. We find them in gene bodies.
par(mfrow = c(1,2))
plot(prom_coverage_all$st_18486 ~ prom_coverage_all$st_18508_dep, xlab="18486", ylab = "18508")
abline(lm(prom_coverage_all$st_18486 ~ prom_coverage_all$st_18508_dep))
plot(prom_coverage_all$st_18486 ~ prom_coverage_all$st_mayer, xlab= "18486", ylab="Mayer")
abline(lm(prom_coverage_all$st_18486 ~ prom_coverage_all$st_mayer))
title("Standard Count Correlatation") 
Compare inclusion and exclusion of promoters
plot(gene_coverage_all$st_18486 ~ prom_coverage_all$st_18486, xlab="gene", ylab = "w/ promoter", main= "Inclusion and exclusion of promoter in counts")
abline(lm(gene_coverage_all$st_18486 ~ prom_coverage_all$st_18486))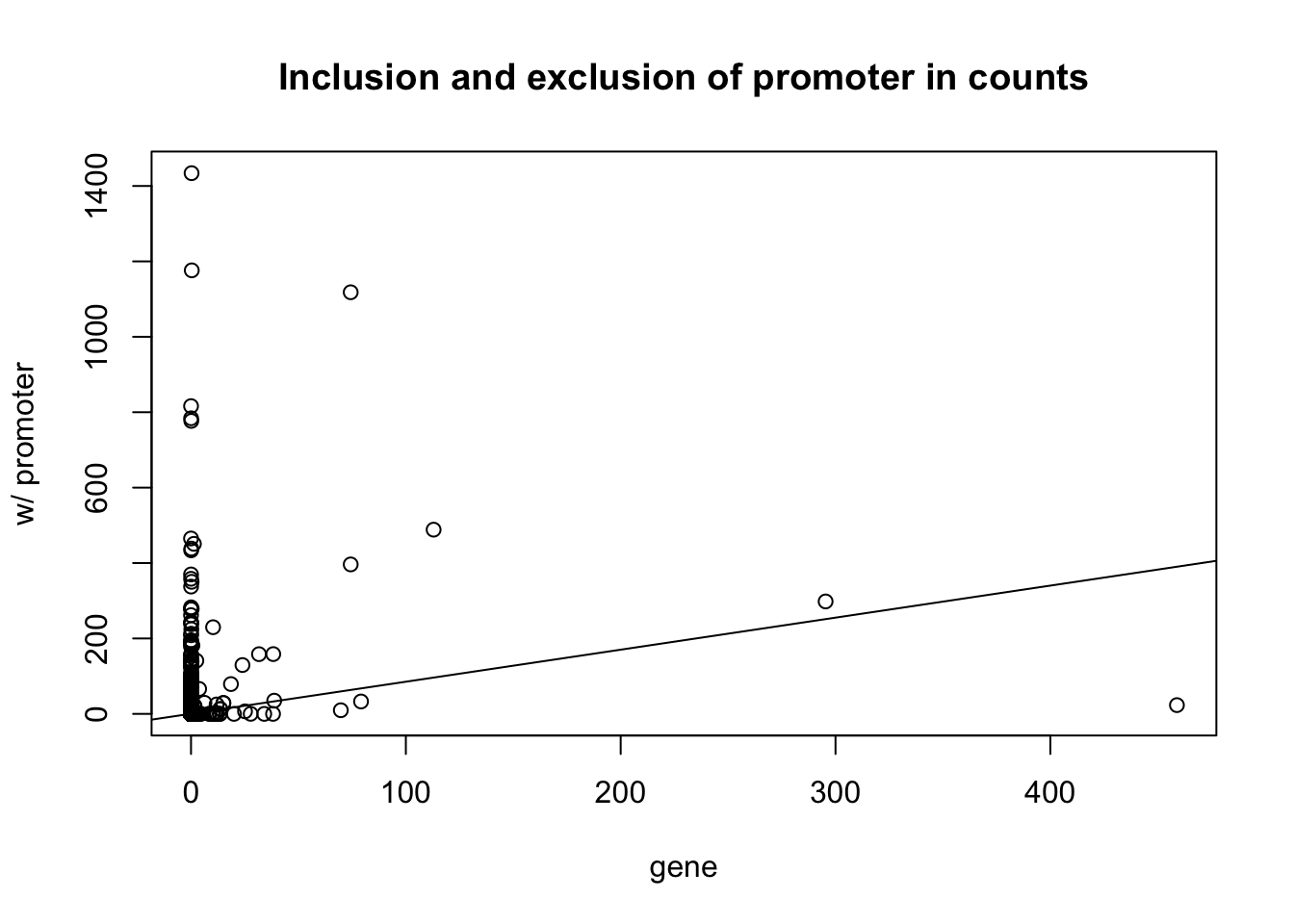
lm(gene_coverage_all$st_18486 ~ prom_coverage_all$st_18486)
Call:
lm(formula = gene_coverage_all$st_18486 ~ prom_coverage_all$st_18486)
Coefficients:
(Intercept) prom_coverage_all$st_18486
0.3291 0.8501 plot(gene_coverage_all$st_18508_dep ~ gene_coverage_all$c_18508_nondep, xlab="dep", ylab = "non_dep", main= "Effect of depletion on gene counts ")
abline(lm(gene_coverage_all$st_18508_dep ~ gene_coverage_all$st_18508_nondep))
lm(gene_coverage_all$st_18508_dep ~ gene_coverage_all$st_18508_nondep)
Call:
lm(formula = gene_coverage_all$st_18508_dep ~ gene_coverage_all$st_18508_nondep)
Coefficients:
(Intercept) gene_coverage_all$st_18508_nondep
0.01254 0.98365 Exclude zeros:
par(mfrow = c(1,2))
prom_no0_18486=prom_coverage_all$st_18486[prom_coverage_all$st_18486!=0]
plot(sort(log(prom_no0_18486), decreasing = TRUE), main="18486 genes and promoters \n without zeros", ylab="log of st. counts")
abline(a= mean(log(prom_no0_18486)), b=0)
gene_no0_18486=gene_coverage_all$st_18486[gene_coverage_all$st_18486 !=0]
plot(sort(log(gene_no0_18486), decreasing = TRUE), main="18486 genes without zeros", ylab="log of st. counts")
abline(a =mean(log(gene_no0_18486)), b=0) Do the same with Mayer
Do the same with Mayer
par(mfrow = c(1,2))
prom_no0_m=prom_coverage_all$st_mayer[prom_coverage_all$st_mayer!=0]
plot(sort(log(prom_no0_m), decreasing = TRUE), main="Mayer genes and promoters \n without zeros", ylab="log of st. counts")
abline(a= mean(log(prom_no0_m)), b=0)
gene_no0_m=gene_coverage_all$st_mayer[gene_coverage_all$st_mayer !=0]
plot(sort(log(gene_no0_m), decreasing = TRUE), main="Mayer genes without zeros", ylab="log of st. counts")
abline(a =mean(log(gene_no0_m)), b=0)
Propotion of detected genes including promoters
Take the sum of the genes that do not equal zero for each set. Make a histogram.
prop_prom_non0_18486= sum(prom_coverage_all$c_18486 !=0) /length(prom_coverage_all$c_18486)
prop_prom_non0_18508dep= sum(prom_coverage_all$c_18508_dep !=0) /length(prom_coverage_all$c_18508_dep)
prop_prom_non0_18508_nondep= sum(prom_coverage_all$c_18508_nondep !=0) /length(prom_coverage_all$c_18508_nondep)
prop_prom_non0_19238= sum(prom_coverage_all$c_19238_dep !=0) /length(prom_coverage_all$c_19238_dep)
prop_prom_non0_mayer= sum(prom_coverage_all$c_mayer !=0) /length(prom_coverage_all$c_mayer)
prop_prom_non0_all= c(prop_prom_non0_18486,prop_prom_non0_18508dep, prop_prom_non0_18508_nondep, prop_prom_non0_19238, prop_prom_non0_mayer)
barplot(prop_prom_non0_all, names=c("18486", "18505 \n dep", "18505 \n non dep", "19238", "Mayer"), ylab= "Proportion of detected promoters", ylim=0:1, xlab= "Library", main = "Proportion of detected promoters")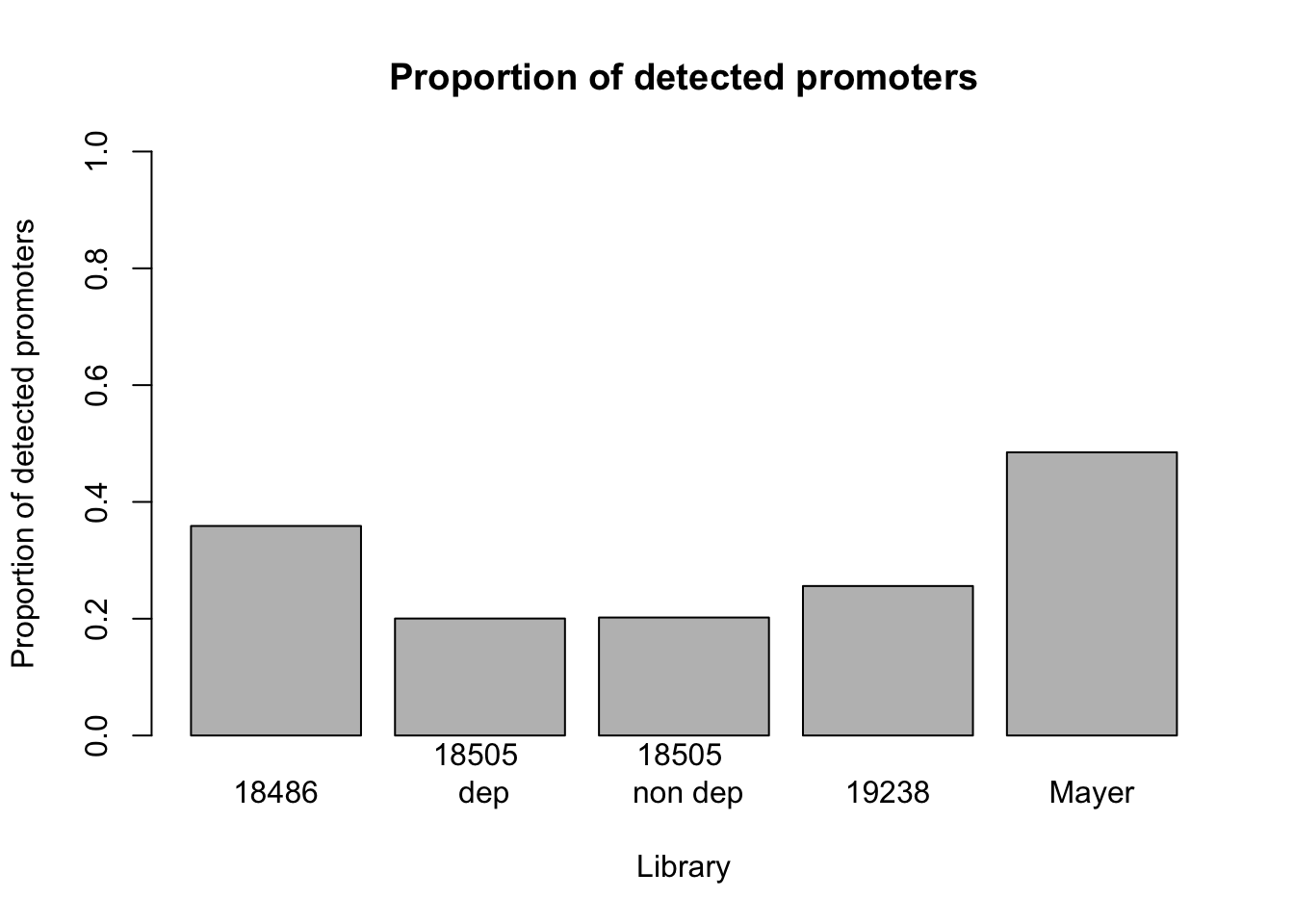
Do this with deduplicated files
The bed files are:
/project2/gilad/briana/Net-seq/Net-seq1/data/bed_dedup/chr
/project2/gilad/briana/Net-seq/data/bed_dedup/chr
This makes the count coverage files for each deduplicated files. gene_dedup_cov.sh
#!/bin/bash
#SBATCH --job-name=gene_dedup_cov
#SBATCH --time=8:00:00
#SBATCH --partition=gilad
#SBATCH --mem=50G
#SBATCH --mail-type=END
cd /project2/gilad/briana/Net-seq/ref_genes
bedtools coverage -counts -a ref_seq_gene_hg19_small_cut -b /project2/gilad/briana/Net-seq/Net-seq1/data/bed_dedup/chr/YG-SP-NET1-18486-dep-2017-10-13_S4_R1_001-sort.dedup.chr.bed > gene_cov_dedup_count/gene_coverage_18486_dedup_count.txt
bedtools coverage -counts -a ref_seq_gene_hg19_small_cut -b /project2/gilad/briana/Net-seq/Net-seq1/data/bed_dedup/chr/YG-SP-NET1-18508-dep-2017-10-13_S2_R1_001-sort.dedup.chr.bed > gene_cov_dedup_count/gene_coverage_18508_dep_dedup_count.txt
bedtools coverage -counts -a ref_seq_gene_hg19_small_cut -b /project2/gilad/briana/Net-seq/Net-seq1/data/bed_dedup/chr/YG-SP-NET1-18508-nondep-2017-10-13_S3_R1_001-sort.dedup.chr.bed > gene_cov_dedup_count/gene_coverage_18508_nondep_dedup_count.txt
bedtools coverage -counts -a ref_seq_gene_hg19_small_cut -b /project2/gilad/briana/Net-seq/Net-seq1/data/bed_dedup/chr/YG-SP-NET1-19238-2017-10-13-S6-R1-001.sort.dedup.chr.bed > gene_cov_dedup_count/gene_coverage_19238_dep_dedup_count.txt
bedtools coverage -counts -a ref_seq_gene_hg19_small_cut -b /project2/gilad/briana/Net-seq/data/bed_dedup/chr/SRR1575922-sort.dedup.chr.bed > gene_cov_dedup_count/gene_coverage_mayer_SRR1575922_dedup_count.txt
The txt files are in /project2/gilad/briana/Net-seq/ref_genes/gene_cov_dedup_count
Input the data:
dedup_gene_cov_18486_count= read.csv("../data/gene_dedup_cov_count/gene_coverage_18486_dedup_count.txt", header=FALSE, sep="\t")
dedup_gene_cov_18508_dep_count= read.csv("../data/gene_dedup_cov_count/gene_coverage_18508_dep_dedup_count.txt", header=FALSE, sep="\t")
dedup_gene_cov_18508_nondep_count= read.csv("../data/gene_dedup_cov_count/gene_coverage_18508_nondep_dedup_count.txt", header=FALSE, sep="\t")
dedup_gene_cov_19238_count= read.csv("../data/gene_dedup_cov_count/gene_coverage_19238_dep_dedup_count.txt", header=FALSE, sep="\t")
dedup_gene_cov_mayer_count= read.csv("../data/gene_dedup_cov_count/gene_coverage_mayer_SRR1575922_dedup_count.txt", header=FALSE, sep="\t")Add col names:
colnames(dedup_gene_cov_18486_count) = c("chr", "start", "end", "name", "score", "strand", "counts")
colnames(dedup_gene_cov_18508_dep_count)= c("chr", "start", "end", "name", "score", "strand", "counts")
colnames(dedup_gene_cov_18508_nondep_count)=c("chr", "start", "end", "name", "score", "strand", "counts")
colnames(dedup_gene_cov_19238_count)= c("chr", "start", "end", "name", "score", "strand", "counts")
colnames(dedup_gene_cov_mayer_count)= c("chr", "start", "end", "name", "score", "strand", "counts")Make dedup all file:
dedup_gene_coverage_all= cbind(dedup_gene_cov_18486_count, dedup_gene_cov_18508_dep_count$counts, dedup_gene_cov_18508_nondep_count$counts, dedup_gene_cov_19238_count$counts, dedup_gene_cov_mayer_count$counts)
colnames(dedup_gene_coverage_all)= c("chr", "start", "end", "name", "score", "strand", "de_18486", "de_18508_dep", "de_18508_nondep", "de_19238_dep", "de_mayer")Standardize by gene length:
#add length column
dedup_gene_coverage_all= mutate(dedup_gene_coverage_all, length=end-start)
#add columns for standard count
dedup_gene_coverage_all= mutate(dedup_gene_coverage_all, st_de_18486=de_18486/length)
dedup_gene_coverage_all= mutate(dedup_gene_coverage_all, st_de_18508_dep= de_18508_dep /length)
dedup_gene_coverage_all= mutate(dedup_gene_coverage_all, st_de_18508_nondep=de_18508_nondep/length)
dedup_gene_coverage_all= mutate(dedup_gene_coverage_all, st_de_19238=de_19238_dep/length)
dedup_gene_coverage_all= mutate(dedup_gene_coverage_all, st_de_mayer=de_mayer/length)Plot these:
plot(sort(log(dedup_gene_coverage_all$st_de_18486 + .1), decreasing=TRUE), ylab="log of dedup gene count standardized by length", xlab="Genes", main="18486")
abline(a=log(mean(dedup_gene_coverage_all$st_de_18486 + .1)),b=0)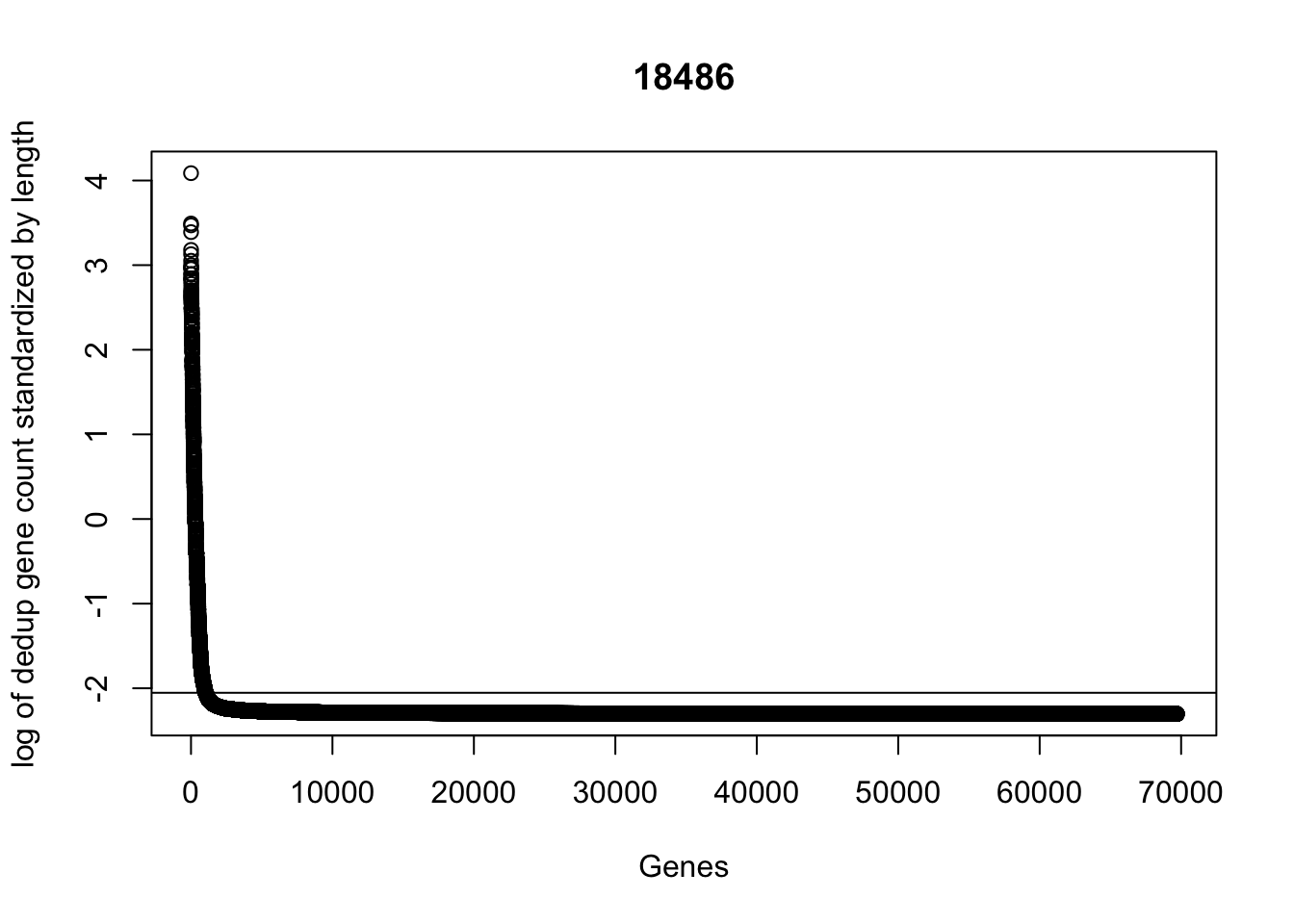
plot(sort(log(dedup_gene_coverage_all$st_de_18508_dep + .1), decreasing=TRUE), ylab="log of dedup gene count standardized by length", xlab="Genes", main="18505 dep")
abline(a=log(mean(dedup_gene_coverage_all$st_de_18508_dep + .1)),b=0)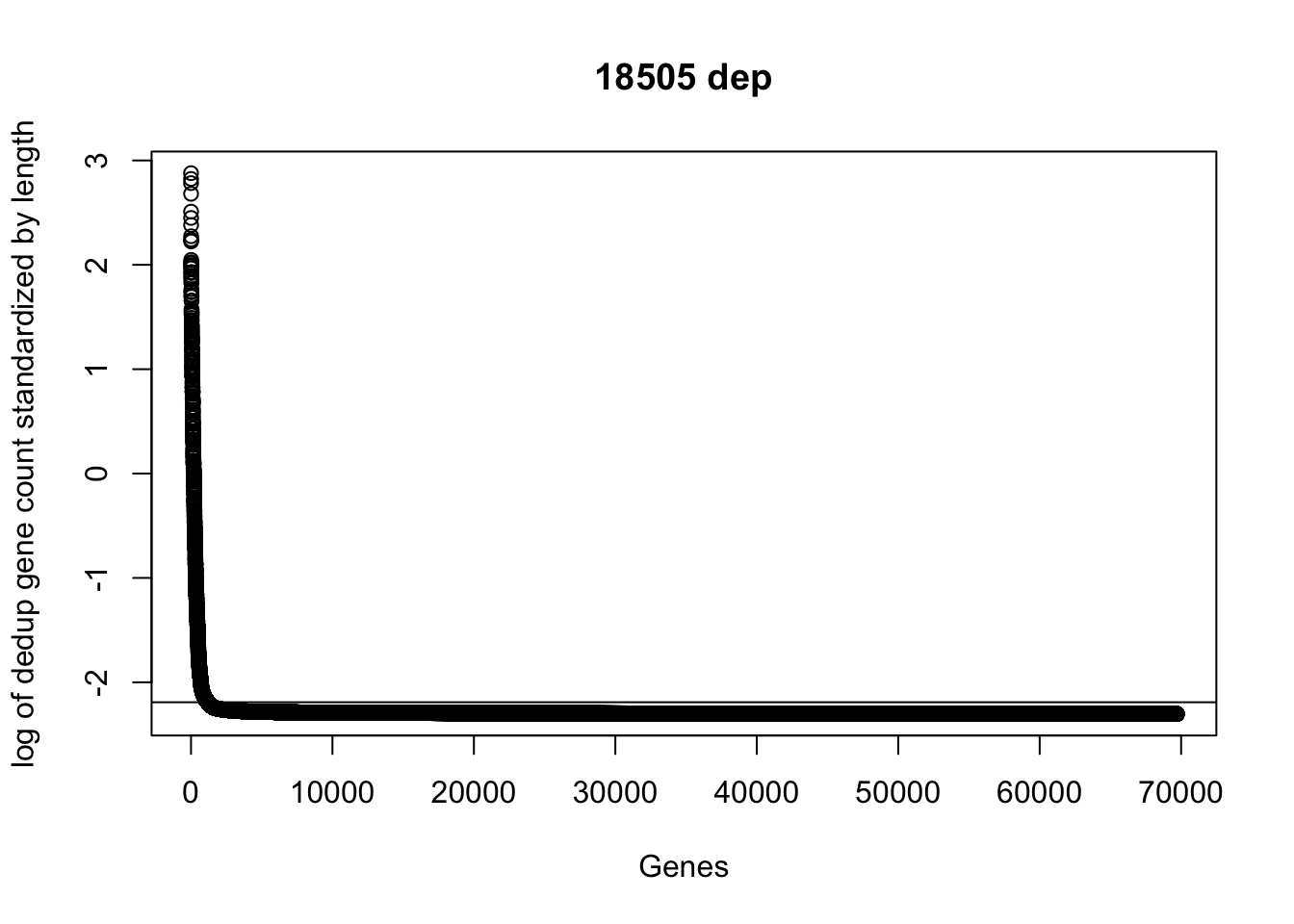
plot(sort(log(dedup_gene_coverage_all$st_de_18508_nondep + .1), decreasing=TRUE), ylab="log of dedup gene count standardized by length", xlab="Genes", main="18505 nondep")
abline(a=log(mean(dedup_gene_coverage_all$st_de_18508_nondep +.1)),b=0)
plot(sort(log(dedup_gene_coverage_all$st_de_19238 + .1), decreasing=TRUE), ylab="log of dedup gene count standardized by length", xlab="Genes", main="19238")
abline(a=log(mean(dedup_gene_coverage_all$st_de_19238 + .1)),b=0)
plot(sort(log(dedup_gene_coverage_all$st_de_mayer + .1), decreasing=TRUE), ylab="log of dedup gene count standardized by length", xlab="Genes", main="Mayer")
abline(a=log(mean(dedup_gene_coverage_all$st_de_mayer + .1)),b=0)
Summaries for gene coverage and deduplicated gene coverage:
#total
summary(gene_coverage_all$st_18486) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.0000 0.0002 0.0007 0.3581 0.0018 1433.9625 summary(gene_coverage_all$st_18508_dep) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.0000 0.0002 0.0006 0.0855 0.0015 405.2400 summary(gene_coverage_all$st_18508_nondep) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.0000 0.0002 0.0007 0.0742 0.0016 335.1200 summary(gene_coverage_all$st_19238) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.0000 0.0001 0.0005 0.1846 0.0012 1552.6600 summary(gene_coverage_all$st_mayer) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.000 0.000 0.001 1.716 0.002 8929.053 #dedup
summary(dedup_gene_coverage_all$st_de_18486) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.00000 0.00011 0.00040 0.02829 0.00096 59.44898 summary(dedup_gene_coverage_all$st_de_18508_dep) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.000000 0.000140 0.000402 0.011789 0.000787 17.682540 summary(dedup_gene_coverage_all$st_de_18508_nondep) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.000000 0.000153 0.000431 0.010702 0.000841 15.730159 summary(dedup_gene_coverage_all$st_de_19238) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.00000 0.00011 0.00033 0.02138 0.00068 57.08163 summary(dedup_gene_coverage_all$st_de_mayer) Min. 1st Qu. Median Mean 3rd Qu. Max.
0.00000 0.00002 0.00065 0.09038 0.00198 80.70492 Look at this with a barplot:
med_18486= c(median(gene_coverage_all$st_18486), median(dedup_gene_coverage_all$st_de_18486))
med_18508_dep=c(median(gene_coverage_all$st_18508_dep), median(dedup_gene_coverage_all$st_de_18508_dep))
med_18508_nondep= c(median(gene_coverage_all$st_18508_nondep), median(dedup_gene_coverage_all$st_de_18508_nondep))
med_19238= c(median(gene_coverage_all$st_19238), median(dedup_gene_coverage_all$st_de_19238))
med_mayer= c(median(gene_coverage_all$st_mayer), median(dedup_gene_coverage_all$st_de_mayer))
median_all=rbind(med_18486, med_18508_dep, med_18508_nondep, med_19238, med_mayer)
colnames(median_all)= c("total", "deduplicated")
#make a barplot
barplot(c(med_18486[1],med_18486[2], med_18508_dep[1], med_18508_dep[2], med_18508_nondep[1], med_18508_nondep[2], med_19238[1], med_19238[2], med_mayer[1], med_mayer[2]), main="Median of standard read counts", col=c("red", "blue","red", "blue","red", "blue","red", "blue","red", "blue"), names= c("18486", "18486", "18508 \ndep", "18508 \ndep", "18508\n nondep", "18508\n nondep", "19238", "19238", "Mayer", "Mayer"), las=2, ylab="Standardized read count", ylim=c(0, .002))
legend("topright",c("reads", "UMI molecules"), col=c("red","blue"), pch=20)
Boxplots:
boxplot(log(dedup_gene_coverage_all$st_de_18486 + .1),log(gene_coverage_all$st_18486 + .1), log(dedup_gene_coverage_all$st_de_18508_dep + .1), log(gene_coverage_all$st_18508_dep + .1), log(dedup_gene_coverage_all$st_de_18508_nondep + .1),log(gene_coverage_all$st_18508_nondep + .1) , log(dedup_gene_coverage_all$st_de_19238 + .1), log(gene_coverage_all$st_19238 + .1), log(dedup_gene_coverage_all$st_de_mayer + .1),log(gene_coverage_all$st_mayer + .1),las = 2, main= "Log of gene counts + .1", names=c("dedup \n18486","18486", "dedup \n 18508 \n dep", " 18508 \n dep","dedup \n 18508 \n nondep","18508 \n nondep","dedup \n 19238", "19238","dedup\n mayer", "mayer"), at =c(1,2,4,5,7,8,10,11,13,14), ylab="log count +.1") Maybe I should make this same plot but remover the zeros:
Maybe I should make this same plot but remover the zeros:
non_zero_dedup_18486= dedup_gene_coverage_all$st_de_18486[dedup_gene_coverage_all$st_de_18486!=0]
non_zero_18486= gene_coverage_all$st_18486[gene_coverage_all$st_18486!=0]
non_zero_dedup_18508_dep= dedup_gene_coverage_all$st_de_18508_dep[dedup_gene_coverage_all$st_de_18508_dep!=0]
non_zero_18508_dep= gene_coverage_all$st_18508_dep[gene_coverage_all$st_18508_dep!=0]
non_zero_dedup_18508_nondep=dedup_gene_coverage_all$st_de_18508_nondep[dedup_gene_coverage_all$st_de_18508_nondep!=0]
non_zero_18508_nondep=gene_coverage_all$st_18508_nondep[gene_coverage_all$st_18508_nondep!=0]
non_zero_dedup_19238=dedup_gene_coverage_all$st_de_19238[dedup_gene_coverage_all$st_de_19238!=0]
non_zero_19238= gene_coverage_all$st_19238[gene_coverage_all$st_19238!=0]
non_zero_dedup_mayer= dedup_gene_coverage_all$st_de_mayer[dedup_gene_coverage_all$st_de_mayer!=0]
non_zero_mayer= gene_coverage_all$st_mayer[gene_coverage_all$st_mayer!=0]
boxplot(log(non_zero_dedup_18486), log(non_zero_18486), log(non_zero_dedup_18508_dep), log(non_zero_18508_dep), log(non_zero_dedup_18508_nondep), log(non_zero_18508_nondep), log(non_zero_dedup_19238), log(non_zero_19238), log(non_zero_dedup_mayer), log(non_zero_mayer), las = 2, main= "Log of non zero gene counts", names=c("dedup \n18486","18486", "dedup \n 18508 \n dep", " 18508 \n dep","dedup \n 18508 \n nondep","18508 \n nondep","dedup \n 19238", "19238","dedup\n mayer", "mayer"), at =c(1,2,4,5,7,8,10,11,13,14), ylab="log count")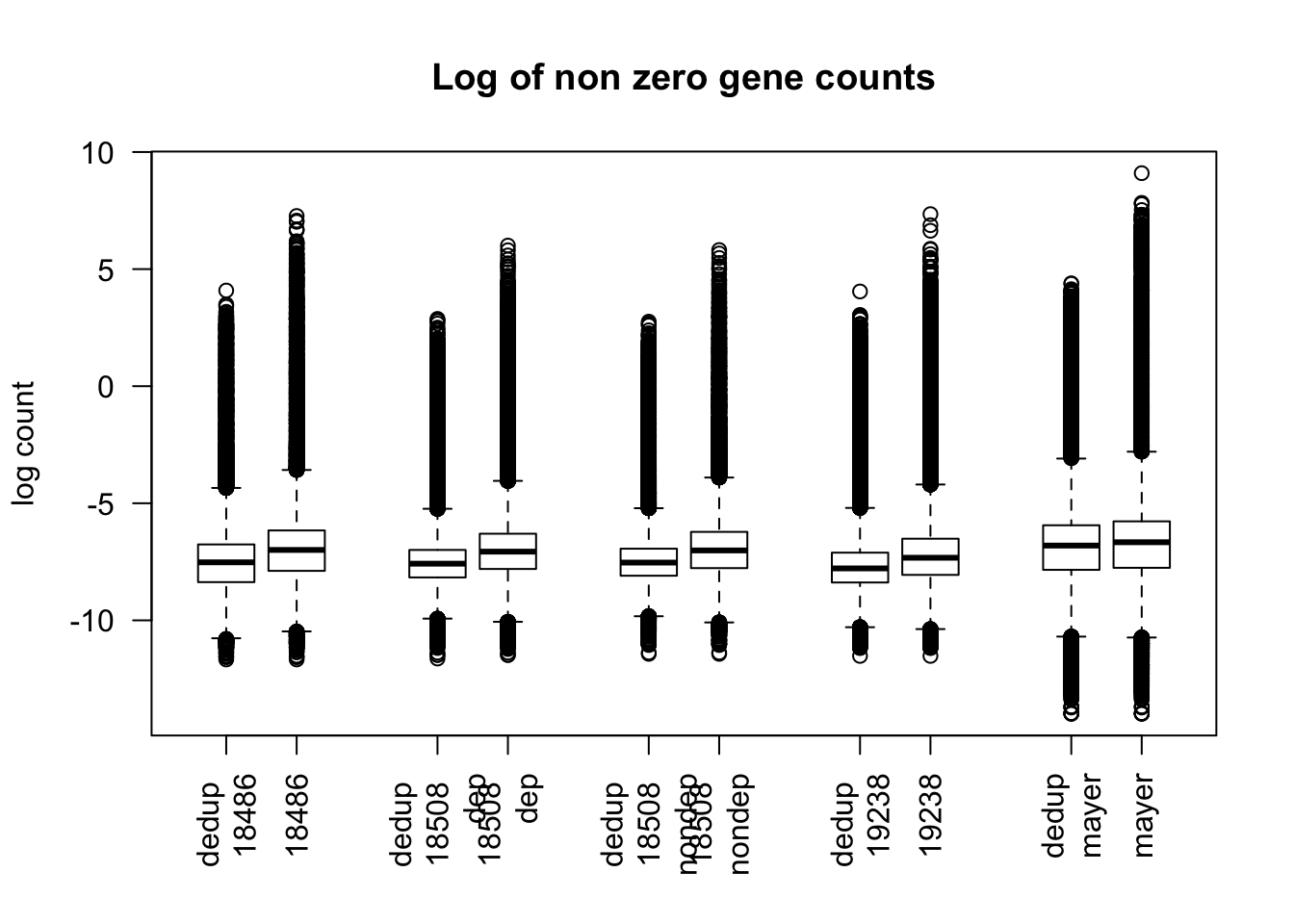
Look at the number of detected genes. This should be the same as the detected genes. The numbers are just smaller per gene:
prop_dedup_gene_non0_18486= sum(dedup_gene_coverage_all$de_18486 != 0) /length(dedup_gene_coverage_all$de_18486)
prop_dedup_gene_non0_18508dep= sum(dedup_gene_coverage_all$de_18508_dep != 0) /length(dedup_gene_coverage_all$de_18508_dep)
prop_dedup_gene_non0_18508_nondep= sum(dedup_gene_coverage_all$de_18508_nondep != 0) /length(dedup_gene_coverage_all$de_18508_nondep)
prop_dedup_gene_non0_19238= sum(dedup_gene_coverage_all$de_19238_dep != 0) /length(dedup_gene_coverage_all$de_19238_dep)
prop_dedup_gene_non0_mayer= sum(dedup_gene_coverage_all$de_mayer != 0) /length(dedup_gene_coverage_all$de_mayer)
barplot( c(prop_gene_non0_18486,prop_gene_non0_18508dep, prop_gene_non0_18508_nondep, prop_gene_non0_19238, prop_gene_non0_mayer) , names=c("18486", "18505 \n dep", "18505 \n non dep", "19238", "Mayer"), ylab= "Proportion of detected genes", ylim=0:1, xlab= "Library", main = "Genes detected in deduplicated libraries")
200 bp windows:
Exon coverage
# exon_coverage_18486_count= read.csv("../data/exon_cov/exon_coverage_18486_count.txt", header=FALSE, sep="\t")
#
# exon_coverage_18508_dep_count= read.csv("../data/exon_cov/exon_coverage_18508_dep_count.txt", header=FALSE, sep="\t")
#
# exon_coverage_18508_nondep_count= read.csv("../data/exon_cov/exon_coverage_18508_nondep_count.txt", header=FALSE, sep="\t")
#
# exon_coverage_19238_dep_count= read.csv("../data/exon_cov/exon_coverage_19238_dep_count.txt", header=FALSE, sep="\t")
#
# exon_coverage_mayer_dep_count = read.csv("../data/exon_cov/exon_coverage_mayer_SRR1575922_count.txt", header=FALSE, sep="\t")# colnames(exon_coverage_mayer_dep_count)=c("chr", "start", "end", "name", "score", "strand", "counts")
# colnames(exon_coverage_19238_dep_count)= c("chr", "start", "end", "name", "score", "strand", "counts")
# colnames(exon_coverage_18508_nondep_count)= c("chr", "start", "end", "name", "score", "strand", "counts")
# colnames(exon_coverage_18508_dep_count)= c("chr", "start", "end", "name", "score", "strand", "counts")
# colnames(exon_coverage_18486_count)= c("chr", "start", "end", "name", "score", "strand", "counts") Coverage with histograms
These are the historgram files I made.
#gene_coverage_18486_dedup= read.csv("../data/gene_coverage_18486_dedup_hist.txt", header=FALSE, sep="\t")
#gene_coverage_18486= read.csv("../data/gene_coverage_18486_hist.txt", head=FALSE, sep="\t")
#gene_coverage_18508_dep= read.csv("../data/gene_coverage_18508_dep_hist.txt", header=FALSE, sep="\t")
#gene_coverage_18508_nondep= read.csv("../data/gene_coverage_18508_nondep_hist.txt", head=FALSE, sep="\t")
#gene_coverage_19238_dep = read.csv("../data/gene_coverage_19238_dep_hist.txt", head=FALSE, sep="\t")
#gene_coverage_mayer_dep = read.csv("../data/gene_coverage_mayer_SRR1575922_hist.txt", head=FALSE, sep="\t")Add column names:
#colnames(gene_coverage_18486_dedup)= c("chr", "start", "end", "name", "score", "strand", "overlap", "bases_non_zero", "lengthA", "frac_A_non_zero_hist")
#colnames(gene_coverage_18486)= c("chr", "start", "end", "name", "score", "strand", "overlap", "bases_non_zero", "lengthA", "frac_A_non_zero_hist")
#colnames(gene_coverage_18508_dep)= c("chr", "start", "end", "name", "score", "strand", "overlap", "bases_non_zero", "lengthA", "frac_A_non_zero_hist")
#colnames(gene_coverage_18508_nondep)= c("chr", "start", "end", "name", "score", "strand", "overlap", "bases_non_zero", "lengthA", "frac_A_non_zero_hist")
#colnames(gene_coverage_19238_dep)= c("chr", "start", "end", "name", "score", "strand", "overlap", "bases_non_zero", "lengthA", "frac_A_non_zero_hist")
#colnames(gene_coverage_mayer_dep)= c("chr", "start", "end", "name", "score", "strand", "overlap", "bases_non_zero", "lengthA", "frac_A_non_zero_hist")Omit NA columns:
# gene_coverage_18486= na.omit(gene_coverage_18486)
# gene_coverage_18486_dedup= na.omit(gene_coverage_18486_dedup)
# gene_coverage_18508_dep= na.omit(gene_coverage_18508_hist_dep)
# gene_coverage_18508_nondep= na.omit(gene_coverage_18508_nondep)
# gene_coverage_19238_dep= na.omit(gene_coverage_19238_dep)
# gene_coverage_mayer_dep= na.omit(gene_coverage_mayer_dep)Session information
sessionInfo()R version 3.4.2 (2017-09-28)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Sierra 10.12.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] bindrcpp_0.2 lattice_0.20-35 ggplot2_2.2.1 dplyr_0.7.4
[5] vioplot_0.2 sm_2.2-5.4
loaded via a namespace (and not attached):
[1] Rcpp_0.12.13 knitr_1.17 bindr_0.1 magrittr_1.5
[5] munsell_0.4.3 colorspace_1.3-2 R6_2.2.2 rlang_0.1.4
[9] plyr_1.8.4 stringr_1.2.0 tools_3.4.2 grid_3.4.2
[13] gtable_0.2.0 git2r_0.19.0 htmltools_0.3.6 lazyeval_0.2.1
[17] yaml_2.1.14 rprojroot_1.2 digest_0.6.12 assertthat_0.2.0
[21] tibble_1.3.4 glue_1.2.0 evaluate_0.10.1 rmarkdown_1.6
[25] labeling_0.3 stringi_1.1.5 compiler_3.4.2 scales_0.5.0
[29] backports_1.1.1 pkgconfig_2.0.1 This R Markdown site was created with workflowr