PAS graph (r markdown)
Mayher
7/26/2019
Last updated: 2020-02-06
Checks: 7 0
Knit directory: apaQTL/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.5.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190411) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: .Rprofile
Untracked: ._.DS_Store
Untracked: .gitignore
Untracked: @
Untracked: GEO_brimittleman/
Untracked: _workflowr.yml
Untracked: analysis/._PASdescriptiveplots.Rmd
Untracked: analysis/._cuttoffPercUsage.Rmd
Untracked: analysis/APApeak_Phenotype_GeneLocAnno.Nuclear.5perc.fc.gz.qqnorm.allChrom
Untracked: analysis/APApeak_Phenotype_GeneLocAnno.Total.5perc.fc.gz.qqnorm.allChrom
Untracked: analysis/QTLexampleplots.Rmd
Untracked: analysis/cuttoffPercUsage.Rmd
Untracked: analysis/eQTLoverlap.Rmd
Untracked: analysis/interpret verify bam.Rmd
Untracked: analysis/interpret_verifybam.Rmd
Untracked: analysis/mergeRNA.Rmd
Untracked: analysis/oldstuffNotNeeded.Rmd
Untracked: analysis/remove_badlines.Rmd
Untracked: analysis/totSpecInNuclear.Rmd
Untracked: analysis/totSpecIncludenotTested.Rmd
Untracked: analysis/totalspec.Rmd
Untracked: apaQTL.Rproj
Untracked: checksumsfastq.txt.gz
Untracked: code/.NascentRNAdtPlotFirstintronicPAS.sh.swp
Untracked: code/._ApaQTL_nominalNonnorm.sh
Untracked: code/._BothFracDTPlotGeneRegions.sh
Untracked: code/._BothFracDTPlotGeneRegions_normalized.sh
Untracked: code/._DistPAS2Sig_RandomIntron.py
Untracked: code/._EandPqtl_perm.sh
Untracked: code/._EandPqtls.sh
Untracked: code/._FC_NucintornUpandDown.sh
Untracked: code/._FC_UTR.sh
Untracked: code/._FC_intornUpandDownsteamPAS.sh
Untracked: code/._FC_nascentseq.sh
Untracked: code/._FC_newPeaks_olddata.sh
Untracked: code/._HMMpermuteTotal.py
Untracked: code/._HmmPermute.py
Untracked: code/._IntronicPASDT.sh
Untracked: code/._LC_samplegroups.py
Untracked: code/._LD_qtl.sh
Untracked: code/._LD_snpsproxy.sh
Untracked: code/._MapAllRBP.sh
Untracked: code/._NascentRNAdtPlot.sh
Untracked: code/._NascentRNAdtPlot3UTRPAS.sh
Untracked: code/._NascentRNAdtPlotExcludeFirstintronicPAS.sh
Untracked: code/._NascentRNAdtPlotNucPAS.sh
Untracked: code/._NascentRNAdtPlotTotPAS.sh
Untracked: code/._NascentRNAdtPlotintronicPAS.sh
Untracked: code/._NascnetRNAdtPlotPAS.sh
Untracked: code/._NetSeq_fourthintronDT.sh
Untracked: code/._NomResfromPASSNP.py
Untracked: code/._NuclearPAS_5per.bed.py
Untracked: code/._NuclearandRNA5samp_dtplots.sh
Untracked: code/._PTTfacetboxplots.R
Untracked: code/._PrematureQTLNominal.sh
Untracked: code/._PrematureQTLPermuted.sh
Untracked: code/._QTL2bed.py
Untracked: code/._QTL2bed_withstrand.py
Untracked: code/._RNAbam2bw.sh
Untracked: code/._RNAseqDTplot.sh
Untracked: code/._Rplots.pdf
Untracked: code/._RunRes2PAS.sh
Untracked: code/._SAF215upbed.py
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilefiltPAS
Untracked: code/._TESplots100bp.sh
Untracked: code/._TESplots150bp.sh
Untracked: code/._TESplots200bp.sh
Untracked: code/._TotalPAS_5perc.bed.py
Untracked: code/._Untitled
Untracked: code/._ZipandTabPheno.sh
Untracked: code/._aAPAqtl_nominal39ind.sh
Untracked: code/._allNucSpecQTLine.py
Untracked: code/._allNucSpecfromNonNorm.py
Untracked: code/._annotatePacBioPASregion.sh
Untracked: code/._annotatedPAS2bed.py
Untracked: code/._apaInPandE.py
Untracked: code/._apaQTLCorrectPvalMakeQQ.R
Untracked: code/._apaQTLCorrectpval_6or7a.R
Untracked: code/._apaQTL_Nominal.sh
Untracked: code/._apaQTL_nominalInclusive.sh
Untracked: code/._apaQTL_nominalv67.sh
Untracked: code/._apaQTL_permuted.sh
Untracked: code/._apaQTL_permuted_test6A7A.sh
Untracked: code/._apainRibo.py
Untracked: code/._assignNucIntonpeak2intronlocs.sh
Untracked: code/._assignTotIntronpeak2intronlocs.sh
Untracked: code/._bam2BW_5primemost.sh
Untracked: code/._bed2saf.py
Untracked: code/._bothFracDTplot1stintron.sh
Untracked: code/._bothFracDTplot4thintron.sh
Untracked: code/._bothFrac_FC.sh
Untracked: code/._callPeaksYL.py
Untracked: code/._changeRibonomQTLres2genename.py
Untracked: code/._changenomQTLres2geneName.py
Untracked: code/._chooseAnno2PAS_pacbio.py
Untracked: code/._chooseAnno2SAF.py
Untracked: code/._chooseSignalSite
Untracked: code/._chooseSignalSite.py
Untracked: code/._closestannotated.sh
Untracked: code/._closestannotated_byfrac.sh
Untracked: code/._cluster.json
Untracked: code/._clusterPAS.json
Untracked: code/._clusterfiltPAS.json
Untracked: code/._codingdms2bed.py
Untracked: code/._config.yaml
Untracked: code/._config2.yaml
Untracked: code/._configOLD.yaml
Untracked: code/._convertNominal2SNPLOC.py
Untracked: code/._convertNominal2SNPloc2Versions.py
Untracked: code/._convertNumeric.py
Untracked: code/._correctNomeqtl.R
Untracked: code/._createPlinkSampfile.py
Untracked: code/._dag.pdf
Untracked: code/._eQTL_switch2snploc.py
Untracked: code/._eQTLgenestestedapa.py
Untracked: code/._encodeRNADTplots.sh
Untracked: code/._extactPAS100meanphyloP.py
Untracked: code/._extractGenotypes.py
Untracked: code/._extractPACmeanPhyloP.py
Untracked: code/._extractseqfromqtlfastq.py
Untracked: code/._fc2leafphen.py
Untracked: code/._fc_filteredPAS6and7As.sh
Untracked: code/._fifteenBPupstreamPAS.py
Untracked: code/._fiftyBPupstreamPAS.py
Untracked: code/._filter5perc.R
Untracked: code/._filter5percPheno.py
Untracked: code/._filterLDsnps.py
Untracked: code/._filterMPPAS.py
Untracked: code/._filterMPPAS_15.py
Untracked: code/._filterMPPAS_15_7As.py
Untracked: code/._filterMPPAS_50.py
Untracked: code/._filterSAFforMP.py
Untracked: code/._filterpeaks.py
Untracked: code/._finalPASbed2SAF.py
Untracked: code/._fix4su304corr.py
Untracked: code/._fix4su604corr.py
Untracked: code/._fix4sukalisto.py
Untracked: code/._fixExandUnexeQTL
Untracked: code/._fixExandUnexeQTL.py
Untracked: code/._fixFChead.py
Untracked: code/._fixFChead_bothfrac.py
Untracked: code/._fixFChead_short.py
Untracked: code/._fixGWAS4Munge.py
Untracked: code/._fixH3k12ac.py
Untracked: code/._fixPASregionSNPs.py
Untracked: code/._fixRNAhead4corr.py
Untracked: code/._fixRNAkalisto.py
Untracked: code/._fix_randomIntron.py
Untracked: code/._fixgroupedtranscript.py
Untracked: code/._fixhead_netseqfc.py
Untracked: code/._getAPAfromanyeQTL.py
Untracked: code/._getApapval4eqtl.py
Untracked: code/._getApapval4eqtl_unexp.py
Untracked: code/._getApapval4eqtl_version67.py
Untracked: code/._getDownstreamIntronNuclear.py
Untracked: code/._getIntronDownstreamPAS.py
Untracked: code/._getIntronUpstreamPAS.py
Untracked: code/._getQTLalleles.py
Untracked: code/._getQTLfastq.sh
Untracked: code/._getUpstreamIntronNuclear.py
Untracked: code/._grouptranscripts.py
Untracked: code/._intersectVCFandupPAS.sh
Untracked: code/._keep5perMAF.py
Untracked: code/._keepSNP_vcf.sh
Untracked: code/._make5percPeakbed.py
Untracked: code/._makeFileID.py
Untracked: code/._makePheno.py
Untracked: code/._makeSAFbothfrac5perc.py
Untracked: code/._makeSNP2rsidfile.py
Untracked: code/._makeeQTLempirical_unexp.py
Untracked: code/._makeeQTLempiricaldist.py
Untracked: code/._makegencondeTSSfile.py
Untracked: code/._mapSSsnps2PAS.sh
Untracked: code/._mergRNABam.sh
Untracked: code/._mergeAllBam.sh
Untracked: code/._mergeAnnotations.sh
Untracked: code/._mergeBW_norm.sh
Untracked: code/._mergeBamNascent.sh
Untracked: code/._mergeByFracBam.sh
Untracked: code/._mergePeaks.sh
Untracked: code/._mnase1stintron.sh
Untracked: code/._mnaseDT_fourthintron.sh
Untracked: code/._namePeaks.py
Untracked: code/._netseqDTplot1stIntron.sh
Untracked: code/._netseqFC.sh
Untracked: code/._nucQTLGWAS.py
Untracked: code/._nucSpecQTLineData.py
Untracked: code/._nucSpeceffectsize.py
Untracked: code/._nucspecnucPASine.py
Untracked: code/._pQTLsotherdata.py
Untracked: code/._pacbioDT.sh
Untracked: code/._pacbioIntronicDT.sh
Untracked: code/._parseBestbamid.py
Untracked: code/._peak2PAS.py
Untracked: code/._peakFC.sh
Untracked: code/._pheno2countonly.R
Untracked: code/._phenoQTLfromlist.py
Untracked: code/._processYRIgen.py
Untracked: code/._pttQTLsinapaQTL.py
Untracked: code/._qtlRegionseq.sh
Untracked: code/._qtlsPvalOppFrac.py
Untracked: code/._quantassign2parsedpeak.py
Untracked: code/._removeXfromHmm.py
Untracked: code/._removeloc_pheno.py
Untracked: code/._riboQTL.sh
Untracked: code/._runCorrectNomEqtl.sh
Untracked: code/._runFixGWAS4Munge.sh
Untracked: code/._runHMMpermuteAPAqtls.sh
Untracked: code/._runHMMpermuteeQTLS.sh
Untracked: code/._runMakeEmpiricaleQTL_unexp.sh
Untracked: code/._runMakeeQTLempirical.sh
Untracked: code/._run_bam2bw_all3prime.sh
Untracked: code/._run_bam2bw_extra3.sh
Untracked: code/._run_bestbamid.sj
Untracked: code/._run_dist2sig_randomintron.sh
Untracked: code/._run_filtersnpLD.sh
Untracked: code/._run_getAPAfromeQTL_version6.7.sh
Untracked: code/._run_getApaPval4eqtl.sh
Untracked: code/._run_getapafromeQTL.py
Untracked: code/._run_getapafromeQTL.sh
Untracked: code/._run_getapapval4eqtl_unexp.sh
Untracked: code/._run_leafcutterDiffIso.sh
Untracked: code/._run_prxySNP.sh
Untracked: code/._run_pttfacetboxplot.sh
Untracked: code/._run_sepUsagephen.sh
Untracked: code/._run_sepgenobychrom.sh
Untracked: code/._run_verifybam.sh
Untracked: code/._selectNominalPvalues.py
Untracked: code/._sepUsagePhen.py
Untracked: code/._sepgenobychrom.py
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._sortindexRNAbam.sh
Untracked: code/._specAPAinE.py
Untracked: code/._submit-snakemakePAS.sh
Untracked: code/._submit-snakemakefiltPAS.sh
Untracked: code/._subsetAPAnotEorPgene.py
Untracked: code/._subsetAPAnotEorPgene_2versions.py
Untracked: code/._subsetApanoteGene.py
Untracked: code/._subsetApanoteGene_2versions.py
Untracked: code/._subsetUnexplainedeQTLs.py
Untracked: code/._subsetVCF_SS.sh
Untracked: code/._subsetVCF_noSSregions.sh
Untracked: code/._subsetVCF_upstreamPAS.sh
Untracked: code/._subset_diffisopheno.py
Untracked: code/._subsetpermAPAwithGenelist.py
Untracked: code/._subsetpermAPAwithGenelist_2versions.py
Untracked: code/._subsetvcf_otherreg.sh
Untracked: code/._subsetvcf_permSS.sh
Untracked: code/._subtrachfiveprimeUTR.sh
Untracked: code/._subtractExons.sh
Untracked: code/._subtractfiveprimeUTR.sh
Untracked: code/._tabixSNPS.sh
Untracked: code/._tenBPupstreamPAS.py
Untracked: code/._test.pdf
Untracked: code/._testVerifyBam.sh
Untracked: code/._totSeceffectsize.py
Untracked: code/._twentyBPupstreamPAS.py
Untracked: code/._utrdms2saf.py
Untracked: code/._vcf2bed.py
Untracked: code/._verifyBam18517N.sh
Untracked: code/._verifyBam18517T.sh
Untracked: code/._verifyBam19128N.sh
Untracked: code/._verifyBam19128T.sh
Untracked: code/._wrap_verifybam.sh
Untracked: code/._writePTTexamplecode.py
Untracked: code/._writePTTexamplecode.sh
Untracked: code/.pversion
Untracked: code/.snakemake/
Untracked: code/1
Untracked: code/APAqtl_nominal.err
Untracked: code/APAqtl_nominal.out
Untracked: code/APAqtl_nominal_39.err
Untracked: code/APAqtl_nominal_39.out
Untracked: code/APAqtl_nominal_inclusive.err
Untracked: code/APAqtl_nominal_inclusive.out
Untracked: code/APAqtl_nominal_nonNorm.err
Untracked: code/APAqtl_nominal_nonNorm.out
Untracked: code/APAqtl_nominal_versions67.err
Untracked: code/APAqtl_nominal_versions67.out
Untracked: code/APAqtl_permuted.err
Untracked: code/APAqtl_permuted.out
Untracked: code/APAqtl_permuted_versions67.err
Untracked: code/APAqtl_permuted_versions67.out
Untracked: code/BothFracDTPlot1stintron.err
Untracked: code/BothFracDTPlot1stintron.out
Untracked: code/BothFracDTPlot4stintron.err
Untracked: code/BothFracDTPlot4stintron.out
Untracked: code/BothFracDTPlotGeneRegions.err
Untracked: code/BothFracDTPlotGeneRegions.out
Untracked: code/BothFracDTPlotGeneRegions_norm.err
Untracked: code/BothFracDTPlotGeneRegions_norm.out
Untracked: code/DistPAS2Sig_RandomIntron.py
Untracked: code/EandPqtl.err
Untracked: code/EandPqtl.out
Untracked: code/EncodeRNADTPlotGeneRegions.err
Untracked: code/EncodeRNADTPlotGeneRegions.out
Untracked: code/FC_NucintronPASupandDown.err
Untracked: code/FC_NucintronPASupandDown.out
Untracked: code/FC_UTR.err
Untracked: code/FC_UTR.out
Untracked: code/FC_intronPASupandDown.err
Untracked: code/FC_intronPASupandDown.out
Untracked: code/FC_nascent.err
Untracked: code/FC_nascentout
Untracked: code/FC_newPAS_olddata.err
Untracked: code/FC_newPAS_olddata.out
Untracked: code/HmmPermute.p
Untracked: code/IntronicPASDT.err
Untracked: code/IntronicPASDT.out
Untracked: code/LD_vcftools.hap.out
Untracked: code/MapAllRBP.sh
Untracked: code/MapRBP.err
Untracked: code/MapRBP.out
Untracked: code/NascentDTPlotGeneRegions.err
Untracked: code/NascentDTPlotGeneRegions.out
Untracked: code/NascentDTPlotPAS.err
Untracked: code/NascentDTPlotPAS.out
Untracked: code/NascentDTPlotPAS_3utr.err
Untracked: code/NascentDTPlotPAS_3utr.out
Untracked: code/NascentDTPlotPAS_firstintron.err
Untracked: code/NascentDTPlotPAS_firstintron.out
Untracked: code/NascentDTPlotPAS_intron.err
Untracked: code/NascentDTPlotPAS_intron.out
Untracked: code/NascentDTPlotPAS_nuc.err
Untracked: code/NascentDTPlotPAS_nuc.out
Untracked: code/NascentDTPlotPAS_tot.err
Untracked: code/NascentDTPlotPAS_tot.out
Untracked: code/Nuclear_example.err
Untracked: code/Nuclear_example.out
Untracked: code/NuclearandRNA5samp_dtplots.sh
Untracked: code/NuclearandRNAFracDTPlotGeneRegions.err
Untracked: code/NuclearandRNAFracDTPlotGeneRegions.out
Untracked: code/PACbioDT.err
Untracked: code/PACbioDT.out
Untracked: code/PACbioDTitronic.err
Untracked: code/PACbioDTitronic.out
Untracked: code/Prematureqtl_nominal.err
Untracked: code/Prematureqtl_nominal.out
Untracked: code/Prematureqtl_permuted.err
Untracked: code/Prematureqtl_permuted.out
Untracked: code/README.md
Untracked: code/RNABam2BW.err
Untracked: code/RNABam2BW.out
Untracked: code/RNAseqDTPlotGeneRegions.err
Untracked: code/RNAseqDTPlotGeneRegions.out
Untracked: code/Rplots.pdf
Untracked: code/TESplots100bp.err
Untracked: code/TESplots100bp.out
Untracked: code/TESplots150bp.err
Untracked: code/TESplots150bp.out
Untracked: code/TESplots200bp.err
Untracked: code/TESplots200bp.out
Untracked: code/Total_example.err
Untracked: code/Total_example.out
Untracked: code/Untitled
Untracked: code/YRI_LCL.vcf.gz
Untracked: code/YRI_LCL_chr1.vcf.gz.log
Untracked: code/YRI_LCL_chr1.vcf.gz.recode.vcf
Untracked: code/annotatedPASregion.err
Untracked: code/annotatedPASregion.out
Untracked: code/apaQTL_nominalInclusive.sh
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assigntotPeak2Intronicregion.err
Untracked: code/assigntotPeak2Intronicregion.out
Untracked: code/bam2bw.err
Untracked: code/bam2bw.out
Untracked: code/bam2bw_5primemost.err
Untracked: code/bam2bw_5primemost.out
Untracked: code/binary_fileset.log
Untracked: code/bothFrac_FC.err
Untracked: code/bothFrac_FC.out
Untracked: code/callSHscripts.txt
Untracked: code/closestannotated.err
Untracked: code/closestannotated.out
Untracked: code/closestannotatedbyfrac.err
Untracked: code/closestannotatedbyfrac.out
Untracked: code/dag.pdf
Untracked: code/dagPAS.pdf
Untracked: code/dagfiltPAS.pdf
Untracked: code/extactPAS100meanphyloP.py
Untracked: code/extractPACmeanPhyloP.py
Untracked: code/fixExandUnexeQTL
Untracked: code/fixGWAS4Munge.py
Untracked: code/fix_randomIntron.py
Untracked: code/fixmunge
Untracked: code/genotypesYRI.gen.proc.keep.vcf.log
Untracked: code/genotypesYRI.gen.proc.keep.vcf.recode.vcf
Untracked: code/getseq100up.err
Untracked: code/getseq100up.out
Untracked: code/grouptranscripts.err
Untracked: code/grouptranscripts.out
Untracked: code/intersectPAS_ssSNPS.err
Untracked: code/intersectPAS_ssSNPS.out
Untracked: code/intersectVCFPAS.err
Untracked: code/intersectVCFPAS.out
Untracked: code/log/
Untracked: code/logs/
Untracked: code/merge53PRIMEbam.err
Untracked: code/merge53PRIMEbam.out
Untracked: code/merge53RNAbam.err
Untracked: code/merge53prime.sh
Untracked: code/merge5RNABam.err
Untracked: code/merge5RNABam.out
Untracked: code/merge5RNAbam.out
Untracked: code/merge5RNAbam.sh
Untracked: code/mergeAnno.err
Untracked: code/mergeAnno.out
Untracked: code/mergeBWnorm.err
Untracked: code/mergeBWnorm.out
Untracked: code/mergeBamNacent.err
Untracked: code/mergeBamNacent.out
Untracked: code/mergeRNAbam.err
Untracked: code/mergeRNAbam.out
Untracked: code/mnaseDTPlot1stintron.err
Untracked: code/mnaseDTPlot1stintron.out
Untracked: code/mnaseDTPlot4thintron.err
Untracked: code/mnaseDTPlot4thintron.out
Untracked: code/netDTPlot4thintron.out
Untracked: code/netseqFC.err
Untracked: code/netseqFC.out
Untracked: code/neyDTPlot4thintron.err
Untracked: code/nucspecinE.py
Untracked: code/plink.log
Untracked: code/prxySNP.err
Untracked: code/prxySNP.out
Untracked: code/pttFacetBoxplots.err
Untracked: code/pttFacetBoxplots.out
Untracked: code/qtlFacetBoxplots.err
Untracked: code/qtlFacetBoxplots.out
Untracked: code/rLD_vcftools.hap.err
Untracked: code/riboqtl.err
Untracked: code/riboqtl.out
Untracked: code/runBestBamID.err
Untracked: code/runCorrectNomeqtl.err
Untracked: code/runCorrectNomeqtl.out
Untracked: code/runFilterLD.err
Untracked: code/runFilterLD.out
Untracked: code/runFixGWAS4Munge.sh
Untracked: code/runHMMpermute.err
Untracked: code/runHMMpermute.out
Untracked: code/runHMMpermuteeQTLs.err
Untracked: code/runHMMpermuteeQTLs.out
Untracked: code/runMakeEmpiricaleQTLs.err
Untracked: code/runMakeEmpiricaleQTLs.out
Untracked: code/runMakeEmpiricaleQTLsunex.err
Untracked: code/runMakeEmpiricaleQTLsunex.out
Untracked: code/run_DistPAS2Sig.err
Untracked: code/run_DistPAS2Sig.out
Untracked: code/run_DistPAS2Sig_intron.err
Untracked: code/run_DistPAS2Sig_intron.out
Untracked: code/run_bam2bw.err
Untracked: code/run_bam2bw.out
Untracked: code/run_bam2bwexta.err
Untracked: code/run_bam2bwexta.out
Untracked: code/run_dist2sig_randomintron.sh
Untracked: code/run_getAPAfromanyeQTL.err
Untracked: code/run_getAPAfromanyeQTL.out
Untracked: code/run_getApaPval4eQTLs.err
Untracked: code/run_getApaPval4eQTLs.out
Untracked: code/run_getApaPval4eQTLsunexplained.err
Untracked: code/run_getApaPval4eQTLsunexplained.out
Untracked: code/run_leafcutter_ds.err
Untracked: code/run_leafcutter_ds.out
Untracked: code/run_sepgenobychrom.err
Untracked: code/run_sepgenobychrom.out
Untracked: code/run_sepusage.err
Untracked: code/run_sepusage.out
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/run_verifybam128N.err
Untracked: code/run_verifybam128N.out
Untracked: code/run_verifybam128T.err
Untracked: code/run_verifybam128T.out
Untracked: code/run_verifybam517N.err
Untracked: code/run_verifybam517N.out
Untracked: code/run_verifybam517T.err
Untracked: code/run_verifybam517T.out
Untracked: code/runprxySNP.err
Untracked: code/runprxySNP.out
Untracked: code/runres2pas.err
Untracked: code/runres2pas.out
Untracked: code/scripts/
Untracked: code/scripts_PAS_500_Lymph/
Untracked: code/seqQTLfastq.err
Untracked: code/seqQTLfastq.out
Untracked: code/seqQTLregion.err
Untracked: code/seqQTLregion.out
Untracked: code/snakePASlog.out
Untracked: code/snakefiltPASlog.out
Untracked: code/sortindexRNABam.err
Untracked: code/sortindexRNABam.out
Untracked: code/specAPAinE.py
Untracked: code/subsetvcf_SS.err
Untracked: code/subsetvcf_SS.out
Untracked: code/subsetvcf_noSS.err
Untracked: code/subsetvcf_noSS.out
Untracked: code/subsetvcf_pas.err
Untracked: code/subsetvcf_pas.out
Untracked: code/subsetvcf_perm.err
Untracked: code/subsetvcf_perm.out
Untracked: code/subsetvcf_rand.err
Untracked: code/subsetvcf_rand.out
Untracked: code/subtract5UTR.err
Untracked: code/subtract5UTR.out
Untracked: code/subtractExons.err
Untracked: code/subtractExons.out
Untracked: code/tabixSNPs.err
Untracked: code/tabixSNPs.out
Untracked: code/test.pdf
Untracked: code/testFix.txt
Untracked: code/test_verifybam.err
Untracked: code/test_verifybam.out
Untracked: code/vcf_keepsnps.err
Untracked: code/vcf_keepsnps.out
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/zipandtabPhen.err
Untracked: code/zipandtabPhen.out
Untracked: data/._.DS_Store
Untracked: data/._MetaDataSequencing.txt
Untracked: data/AnnotatedPAS/
Untracked: data/ApaByEgene/
Untracked: data/ApaByPgene/
Untracked: data/BadLines/
Untracked: data/BaseComp/
Untracked: data/Battle_pQTL/
Untracked: data/CheckSums/
Untracked: data/CompareOldandNew/
Untracked: data/DTmatrix/
Untracked: data/DiffIso/
Untracked: data/EncodeRNA/
Untracked: data/ExampleQTLPlots/
Untracked: data/ExampleQTLPlots_update/
Untracked: data/ExpressionIndependentapaQTLs.txt
Untracked: data/FiveMergedBW/
Untracked: data/FiveMergedBam/
Untracked: data/FlaggedPAS/
Untracked: data/GWAS_overlap/
Untracked: data/GeuvadisRNA/
Untracked: data/HMMqtls/
Untracked: data/LDSR_annotations/
Untracked: data/Li_eQTLs/
Untracked: data/NMD/
Untracked: data/NascentRNA/
Untracked: data/NucSpeceQTLeffect/
Untracked: data/PAS/
Untracked: data/PAS_postFlag/
Untracked: data/PolyA_DB/
Untracked: data/PreTerm_pheno/
Untracked: data/PrematureQTLNominal/
Untracked: data/PrematureQTLPermuted/
Untracked: data/QTLGenotypes/
Untracked: data/QTLoverlap/
Untracked: data/QTLoverlap_inclusive/
Untracked: data/QTLoverlap_nonNorm/
Untracked: data/README.md
Untracked: data/RNAseq/
Untracked: data/Reads2UTR/
Untracked: data/SNPinSS/
Untracked: data/SignalSiteFiles/
Untracked: data/TF_motifdisruption/
Untracked: data/TSS/
Untracked: data/ThirtyNineIndQtl_nominal/
Untracked: data/Version15bp6As/
Untracked: data/Version15bp7As/
Untracked: data/apaQTLNominal/
Untracked: data/apaQTLNominal_4pc/
Untracked: data/apaQTLNominal_inclusive/
Untracked: data/apaQTLPermuted/
Untracked: data/apaQTLPermuted_4pc/
Untracked: data/apaQTLs/
Untracked: data/assignedPeaks/
Untracked: data/assignedPeaks_15Up/
Untracked: data/bam/
Untracked: data/bam_clean/
Untracked: data/bam_waspfilt/
Untracked: data/bed_10up/
Untracked: data/bed_clean/
Untracked: data/bed_clean_sort/
Untracked: data/bed_waspfilter/
Untracked: data/bedsort_waspfilter/
Untracked: data/bothFrac_FC/
Untracked: data/bw/
Untracked: data/bw_norm/
Untracked: data/eCLip/
Untracked: data/eQTLs/
Untracked: data/exampleQTLs/
Untracked: data/fastq/
Untracked: data/filterPeaks/
Untracked: data/fourSU/
Untracked: data/h3k27ac/
Untracked: data/highdiffsiggenes.txt
Untracked: data/inclusivePeaks/
Untracked: data/inclusivePeaks_FC/
Untracked: data/intronRNAratio/
Untracked: data/intron_analysis/
Untracked: data/locusZoom/
Untracked: data/mergedBG/
Untracked: data/mergedBW_byfrac/
Untracked: data/mergedBW_norm/
Untracked: data/mergedBam/
Untracked: data/mergedbyFracBam/
Untracked: data/molPhenos/
Untracked: data/molQTLs/
Untracked: data/motifdistrupt/
Untracked: data/nPAS/
Untracked: data/netseq/
Untracked: data/nonNorm_pheno/
Untracked: data/nuc_10up/
Untracked: data/nuc_10upclean/
Untracked: data/oldPASfiles/
Untracked: data/overlapeQTL_try2/
Untracked: data/overlapeQTLs/
Untracked: data/pQTLoverlap/
Untracked: data/pacbio/
Untracked: data/peakCoverage/
Untracked: data/peaks_5perc/
Untracked: data/phenotype/
Untracked: data/phenotype_5perc/
Untracked: data/phenotype_inclusivePAS/
Untracked: data/phylop/
Untracked: data/pttQTL/
Untracked: data/pttQTLplots/
Untracked: data/sigDiffGenes.txt
Untracked: data/sort/
Untracked: data/sort_clean/
Untracked: data/sort_waspfilter/
Untracked: data/twoMech/
Untracked: data/vareQTLvarAPAqtl/
Untracked: data/verifyBAM/
Untracked: data/verifyBAM_full/
Untracked: nohup.out
Untracked: output/._.DS_Store
Untracked: output/._AverageDiffHeatmap.Nuclear.png
Untracked: output/._AverageDiffHeatmap.Total.png
Untracked: output/._GeneswithAPApotential.png
Untracked: output/._GeneswithAPApotentialAllPAS.png
Untracked: output/._PASlocation.png
Untracked: output/._SignalSitePlot.png
Untracked: output/._meanCorrelationPhenotypes.svg
Untracked: output/._qqplot_Nuclear_APAperm.png
Untracked: output/._qqplot_Nuclear_APAperm_4pc.png
Untracked: output/._qqplot_Total_APAperm.png
Untracked: output/._qqplot_Total_APAperm_4pc.png
Untracked: output/AverageDiffHeatmap.Nuclear.png
Untracked: output/AverageDiffHeatmap.Total.png
Untracked: output/GeneswithAPApotential.png
Untracked: output/GeneswithAPApotentialAllPAS.png
Untracked: output/PASlocation.png
Untracked: output/SignalSitePlot.png
Untracked: output/SignalSitePlotbyLoc.png
Untracked: output/dtPlots/
Untracked: output/fastqc/
Untracked: output/meanCorrelationPhenotypes.svg
Untracked: output/newnuc.png
Untracked: output/newtot.png
Untracked: output/oldnuc.png
Untracked: output/oldtot.png
Untracked: output/qqplot_Nuclear_APAperm.png
Untracked: output/qqplot_Nuclear_APAperm_4pc.png
Untracked: output/qqplot_Total_APAperm.png
Untracked: output/qqplot_Total_APAperm_4pc.png
Untracked: run_verifybam517N.err
Untracked: run_verifybam517N.out
Unstaged changes:
Modified: analysis/NuclearSpecIncludeNotTested.Rmd
Modified: analysis/PASdescriptiveplots.Rmd
Modified: analysis/RNAbinding.Rmd
Modified: analysis/Readdistagainstfeatures.Rmd
Modified: analysis/TSS.Rmd
Modified: analysis/decayAndStability.Rmd
Modified: analysis/nascenttranscription.Rmd
Modified: analysis/nucSpecinEQTLs.Rmd
Modified: analysis/overlapapaqtlsandeqtls.Rmd
Modified: analysis/pQTLexampleplot.Rmd
Modified: analysis/propeQTLs_explained.Rmd
Modified: analysis/version15bpfilter.Rmd
Modified: code/DistPAS2Sig.py
Modified: code/Script4NuclearQTLexamples.sh
Modified: code/Script4TotalQTLexamples.sh
Modified: code/apaQTLsnake.err
Modified: code/run_qtlFacetBoxplots.sh
Deleted: code/test.txt
Deleted: reads_graphs.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 1c54594 | brimittleman | 2020-02-07 | fix size |
| html | 6aa6a5f | brimittleman | 2020-02-07 | Build site. |
| Rmd | 4a2b11f | brimittleman | 2020-02-07 | change size 1 |
| html | 7477dd7 | brimittleman | 2020-02-06 | Build site. |
| Rmd | 8c10277 | brimittleman | 2020-02-06 | add code for LD and fix double y axis |
| html | 8188b97 | brimittleman | 2019-09-23 | Build site. |
| Rmd | 33f15fd | brimittleman | 2019-09-23 | ggplots for pltos |
| html | bb2fbab | brimittleman | 2019-09-11 | Build site. |
| Rmd | 5ad175a | brimittleman | 2019-09-11 | udate pas graphs and asdd total |
| html | 3b25860 | brimittleman | 2019-09-04 | Build site. |
| Rmd | 5984fad | brimittleman | 2019-09-04 | update for new filter condition |
| html | 00d73e3 | brimittleman | 2019-07-31 | Build site. |
| html | 15fbb3c | brimittleman | 2019-07-31 | Build site. |
| Rmd | aa1ad85 | brimittleman | 2019-07-31 | add pdf for figrues |
| html | d13025b | brimittleman | 2019-07-31 | Build site. |
| Rmd | e2ff61a | brimittleman | 2019-07-31 | add mayher plot to site |
| Rmd | 64e6257 | GitHub | 2019-07-31 | Added new file for PAS graphs |
I first install all of the packages and librarys that will be necessary for the formation of these graphs. Lattice Extra in particular has the “doubleYScale()” function that helps me a lot when making these plots.
The current data frame has two columns - ID, and meanUsage. ID is a very long string containing a lot of currently irrelevant information. In this step, I am trying to solely get the type of the PAS from the ID column in the data frame along with its respective mean usage. To do this, I first read in the data frame. I then split the ID column into 5 columns, each for it’s respective type of information. Though I only need “type” it is easier to access the other information if needed in the future. Then, I create a new data frame containing “type” and “mean usage”.
#reading in the data frame
df <- read.delim("../data/PAS/NuclearPASMeanUsage.txt")
#splitting the ID column
sep <-
separate(data = df, col = ID, into = c("chr", "start","end", "thing", "peak"), sep = "\\:|\\_\\+\\_|\\_\\-\\_", remove = TRUE, convert = FALSE, extra = "warn", fill = "warn")
sep <-
separate(data = sep, col = thing, into = c("thing", "type"), sep = "\\_", remove = TRUE, convert = FALSE, extra = "warn", fill = "warn")Warning: Expected 2 pieces. Additional pieces discarded in 3 rows [12630,
12631, 12632].#deleting extraneous information (everything except type and mean usage)
keeps <- c("type","meanUsage")
total <- sep[keeps]Now that I have the information I want, I have to seperate it based on type. Here, I create 5 data frames that contain only the mean usage for each type, as well as the one for total. I then convert each data frame to a data matrix, which changes the type from a list to a double, allowing it to be put in a plot.
#get the meanUsage per type in a data frame
utr3 <- subset(total, type == "utr3", select = c(meanUsage))
utr5 <- subset(total, type == "utr5", select = c(meanUsage))
end <- subset(total, type == "end", select = c(meanUsage))
cds <- subset(total, type == "cds", select = c(meanUsage))
intron <- subset(total, type == "intron", select = c(meanUsage))
#then, do the same for total
total <- total["meanUsage"]
#convert the data frame to a data matrix so it can be used in a plot
utr3_graph <- data.matrix(utr3)
utr5_graph <- data.matrix(utr5)
end_graph <- data.matrix(end)
cds_graph <- data.matrix(cds)
intron_graph <- data.matrix(intron)
total_graph <- data.matrix(total)Here, I prepare for the next step by initializing 6 data frames, one for each type of PAS and one for the total data. Because I want meanUsage cutoff (number of values above a specific cutoff), instead of meanUsage itself, I need this extra step.
cutoff_utr3 <- data.frame("cutoff numbers" = c(0,0,0,0,0,0,0,0,0,0))
cutoff_utr5 <- data.frame("cutoff numbers" = c(0,0,0,0,0,0,0,0,0,0))
cutoff_end <- data.frame("cutoff numbers" = c(0,0,0,0,0,0,0,0,0,0))
cutoff_cds <- data.frame("cutoff numbers" = c(0,0,0,0,0,0,0,0,0,0))
cutoff_intron <- data.frame("cutoff numbers" = c(0,0,0,0,0,0,0,0,0,0))
cutoff_total <- data.frame("cutoff" = c(0,0,0,0,0,0,0,0,0,0))In this step, I take each data matrix, and insert frequencies into the cutoff data frames. For example, if there is a meanUsage greater than 0.4, the cutoff for 0.4, 0.3, 0.2, and 0.1 will increase by one. I do this by using a for loop, and a lot of if statements per for loop to sift the mean usages in their correct “bins”. I do this for all 6 data sets.
for (i in utr3_graph){
if(i >0.1){
cutoff_utr3[1,]<- cutoff_utr3[1,] +1
}
if(i>0.2) {
cutoff_utr3[2,]<- cutoff_utr3[2,] +1
}
if (i>0.3) {
cutoff_utr3[3,]<- cutoff_utr3[3,] +1
}
if(i>0.4) {
cutoff_utr3[4,]<- cutoff_utr3[4,] +1
}
if(i>0.5) {
cutoff_utr3[5,]<- cutoff_utr3[5,] +1
}
if(i>0.6) {
cutoff_utr3[6,]<- cutoff_utr3[6,] +1
}
if(i>0.7) {
cutoff_utr3[7,]<- cutoff_utr3[7,] +1
}
if(i>0.8) {
cutoff_utr3[8,]<- cutoff_utr3[8,] +1
}
if(i>0.9) {
cutoff_utr3[9,]<- cutoff_utr3[9,] +1
}
if(i>=1.0) {
cutoff_utr3[10,]<- cutoff_utr3[10,] +1
}
}
for (i in utr5_graph){
if(i >0.1){
cutoff_utr5[1,]<- cutoff_utr5[1,] +1
}
if(i>0.2) {
cutoff_utr5[2,]<- cutoff_utr5[2,] +1
}
if (i>0.3) {
cutoff_utr5[3,]<- cutoff_utr5[3,] +1
}
if(i>0.4) {
cutoff_utr5[4,]<- cutoff_utr5[4,] +1
}
if(i>0.5) {
cutoff_utr5[5,]<- cutoff_utr5[5,] +1
}
if(i>0.6) {
cutoff_utr5[6,]<- cutoff_utr5[6,] +1
}
if(i>0.7) {
cutoff_utr5[7,]<- cutoff_utr5[7,] +1
}
if(i>0.8) {
cutoff_utr5[8,]<- cutoff_utr5[8,] +1
}
if(i>0.9) {
cutoff_utr5[9,]<- cutoff_utr5[9,] +1
}
if(i>=1.0) {
cutoff_utr5[10,]<- cutoff_utr5[10,] +1
}
}
for (i in end_graph){
if(i >0.1){
cutoff_end[1,]<- cutoff_end[1,] +1
}
if(i>0.2) {
cutoff_end[2,]<- cutoff_end[2,] +1
}
if (i>0.3) {
cutoff_end[3,]<- cutoff_end[3,] +1
}
if(i>0.4) {
cutoff_end[4,]<- cutoff_end[4,] +1
}
if(i>0.5) {
cutoff_end[5,]<- cutoff_end[5,] +1
}
if(i>0.6) {
cutoff_end[6,]<- cutoff_end[6,] +1
}
if(i>0.7) {
cutoff_end[7,]<- cutoff_end[7,] +1
}
if(i>0.8) {
cutoff_end[8,]<- cutoff_end[8,] +1
}
if(i>0.9) {
cutoff_end[9,]<- cutoff_end[9,] +1
}
if(i>=1.0) {
cutoff_end[10,]<- cutoff_end[10,] +1
}
}
for (i in cds_graph){
if(i >0.1){
cutoff_cds[1,]<- cutoff_cds[1,] +1
}
if(i>0.2) {
cutoff_cds[2,]<- cutoff_cds[2,] +1
}
if (i>0.3) {
cutoff_cds[3,]<- cutoff_cds[3,] +1
}
if(i>0.4) {
cutoff_cds[4,]<- cutoff_cds[4,] +1
}
if(i>0.5) {
cutoff_cds[5,]<- cutoff_cds[5,] +1
}
if(i>0.6) {
cutoff_cds[6,]<- cutoff_cds[6,] +1
}
if(i>0.7) {
cutoff_cds[7,]<- cutoff_cds[7,] +1
}
if(i>0.8) {
cutoff_cds[8,]<- cutoff_cds[8,] +1
}
if(i>0.9) {
cutoff_cds[9,]<- cutoff_cds[9,] +1
}
if(i>=1.0) {
cutoff_cds[10,]<- cutoff_cds[10,] +1
}
}
for (i in intron_graph){
if(i >0.1){
cutoff_intron[1,]<- cutoff_intron[1,] +1
}
if(i>0.2) {
cutoff_intron[2,]<- cutoff_intron[2,] +1
}
if (i>0.3) {
cutoff_intron[3,]<- cutoff_intron[3,] +1
}
if(i>0.4) {
cutoff_intron[4,]<- cutoff_intron[4,] +1
}
if(i>0.5) {
cutoff_intron[5,]<- cutoff_intron[5,] +1
}
if(i>0.6) {
cutoff_intron[6,]<- cutoff_intron[6,] +1
}
if(i>0.7) {
cutoff_intron[7,]<- cutoff_intron[7,] +1
}
if(i>0.8) {
cutoff_intron[8,]<- cutoff_intron[8,] +1
}
if(i>0.9) {
cutoff_intron[9,]<- cutoff_intron[9,] +1
}
if(i>=1.0) {
cutoff_intron[10,]<- cutoff_intron[10,] +1
}
}
for (i in total_graph){
if(i >0.1){
cutoff_total[1,]<- cutoff_total[1,] +1
}
if(i>0.2) {
cutoff_total[2,]<- cutoff_total[2,] +1
}
if (i>0.3) {
cutoff_total[3,]<- cutoff_total[3,] +1
}
if(i>0.4) {
cutoff_total[4,]<- cutoff_total[4,] +1
}
if(i>0.5) {
cutoff_total[5,]<- cutoff_total[5,] +1
}
if(i>0.6) {
cutoff_total[6,]<- cutoff_total[6,] +1
}
if(i>0.7) {
cutoff_total[7,]<- cutoff_total[7,] +1
}
if(i>0.8) {
cutoff_total[8,]<- cutoff_total[8,] +1
}
if(i>0.9) {
cutoff_total[9,]<- cutoff_total[9,] +1
}
if(i>=1.0) {
cutoff_total[10,]<- cutoff_total[10,] +1
}
} I now make data frames that contain the proportion of the specific type of PAS with respect to the total. I do this by dividing the frequency of the mean usage per cutoff of the type, by the same in the total.
utr3_prop <- cutoff_utr3/cutoff_total
utr5_prop <- cutoff_utr5/cutoff_total
end_prop <- cutoff_end/cutoff_total
cds_prop <- cutoff_cds/cutoff_total
intron_prop <- cutoff_intron/cutoff_totalHere, I create a data frame called “breaks_new” that has all of the mean usage cutoffs I used for the other data. I then combined this data frame with the proportion to create the data frames needed for all of the plots.
breaks_new <- data.frame(xval = c(0.10, 0.20, 0.30, 0.40, 0.50, 0.60, 0.70, 0.80, 0.90, 1.00))
totalMeanUsageFreq <- data.frame(breaks_new, cutoff_total)
Utr3prop <- data.frame(breaks_new, utr3_prop)
Utr5prop <- data.frame(breaks_new, utr5_prop)
endprop <- data.frame(breaks_new, end_prop)
cdsprop <- data.frame(breaks_new, cds_prop)
intronprop <- data.frame(breaks_new, intron_prop)Here, I initialixe the objects, which have all of the information in them, such as the type of plots, y axis label, and scale. The first object (obj1), is the one for the total frequency, which will be one side for the double Y axis plot.
obj1 <- xyplot(cutoff ~ xval, totalMeanUsageFreq,
ylab = "Number of Nuclear PAS", xlab = "meanUsage cutoff", col.lab = "black")
obj2 <- xyplot(cutoff.numbers ~ xval, Utr3prop,
panel = function(...)panel.xyplot(type = "l",lty = 2,grid=TRUE,...), ylab = "Proportion in 3' UTR", scales = list(y=list(limits=c(0,1))))
obj3 <- xyplot(cutoff.numbers ~ xval, Utr5prop,
panel = function(...)panel.xyplot(type = "l",lty = 2,grid=TRUE,...), ylab = "Proportion in 5' UTR", scales = list(y=list(limits=c(0,1))))
obj4 <- xyplot(cutoff.numbers ~ xval, endprop,
panel = function(...)panel.xyplot(type = "l",lty = 2,grid=TRUE,...), ylab = "Proportion Downstream", scales = list(y=list(limits=c(0,1))))
obj5 <- xyplot(cutoff.numbers ~ xval, cdsprop,
panel = function(...)panel.xyplot(type = "l",lty = 2,grid=TRUE,...), ylab = "Proportion in Coding", scales = list(y=list(limits=c(0,1))))
obj6 <- xyplot(cutoff.numbers ~ xval, intronprop,
panel = function(...)panel.xyplot(type = "l",lty = 2,grid=TRUE,...), ylab = "Proportion Intronic", scales = list(y=list(limits=c(0,1))))And lastly, I create the final, double Y axis plots
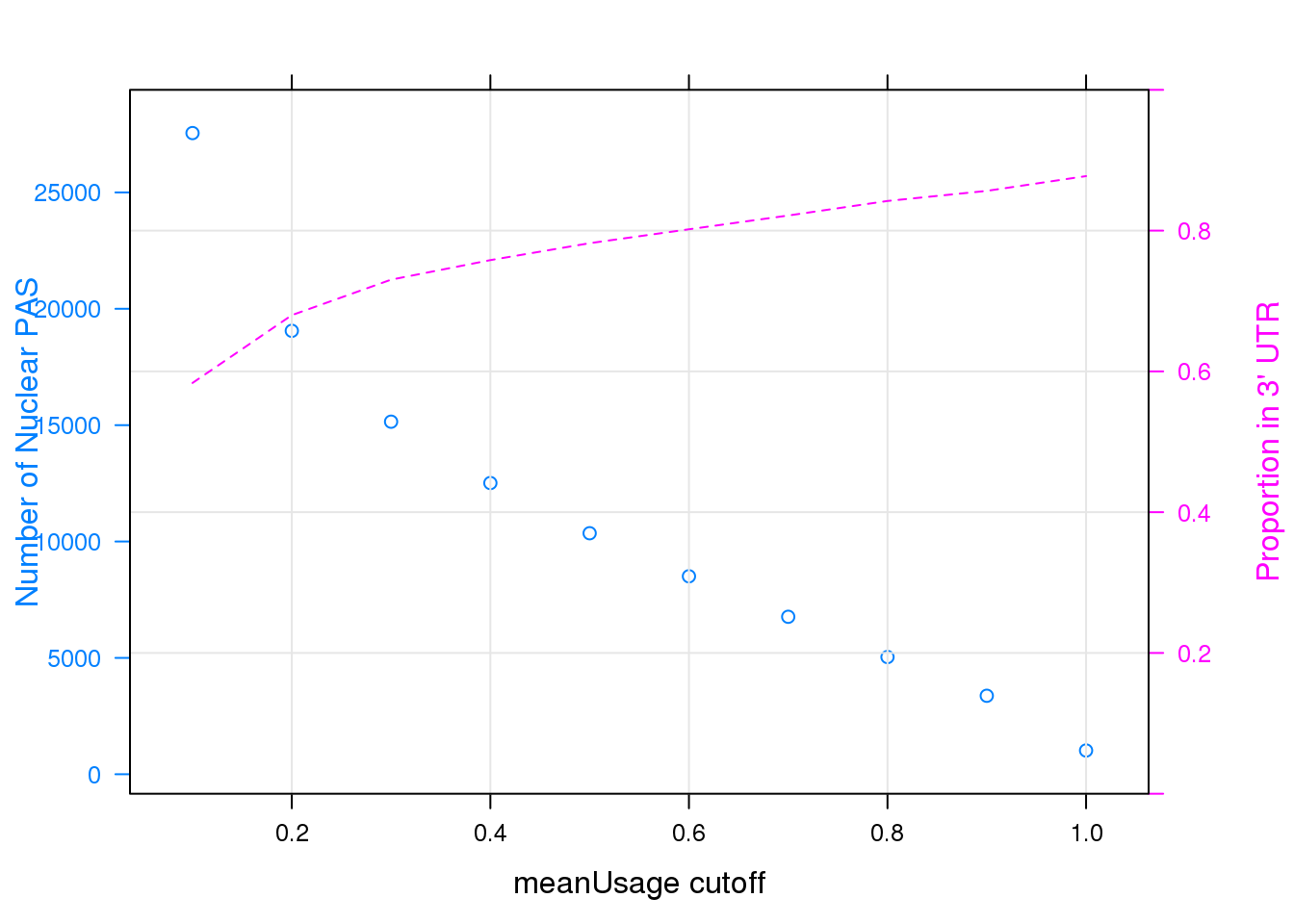
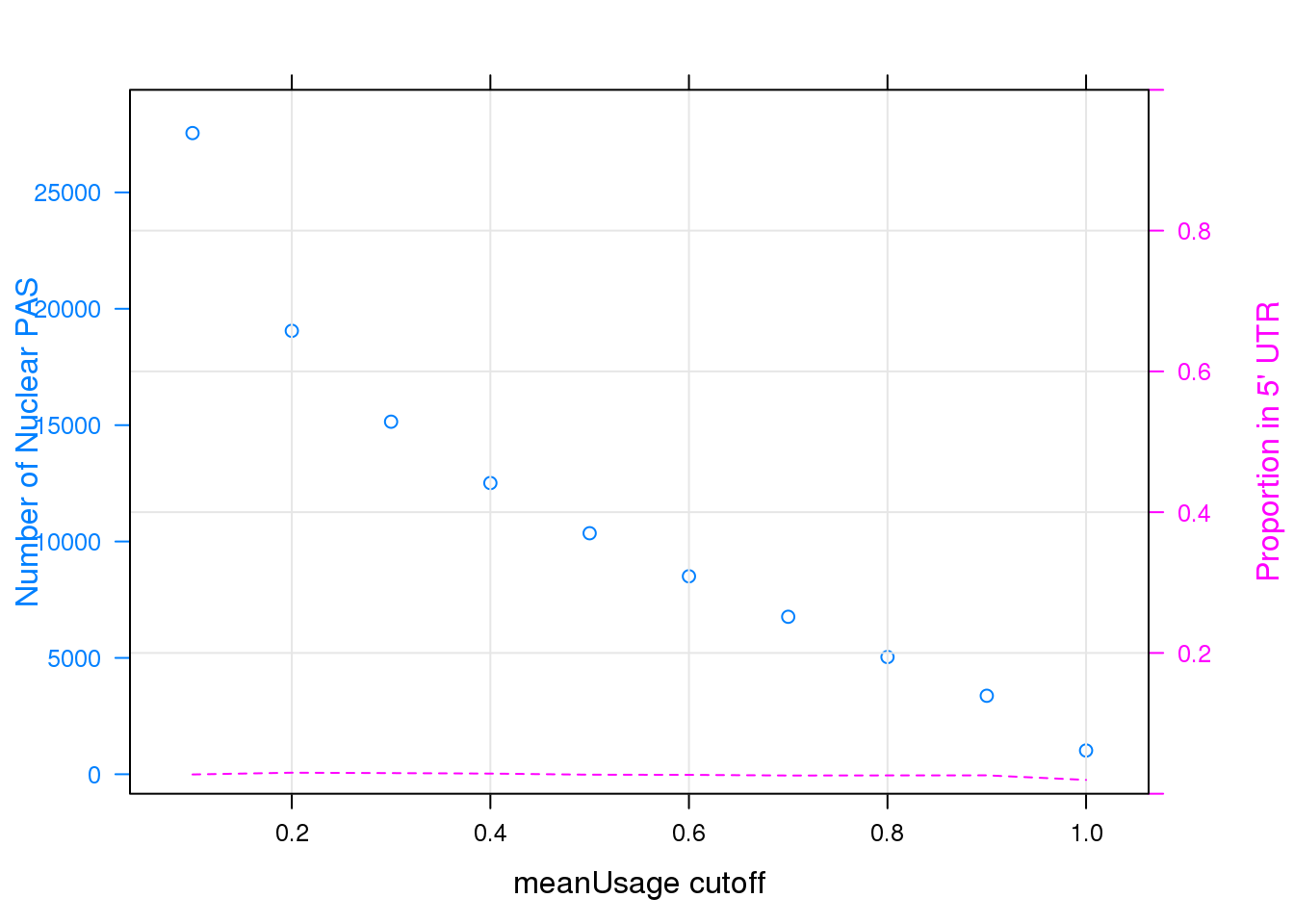
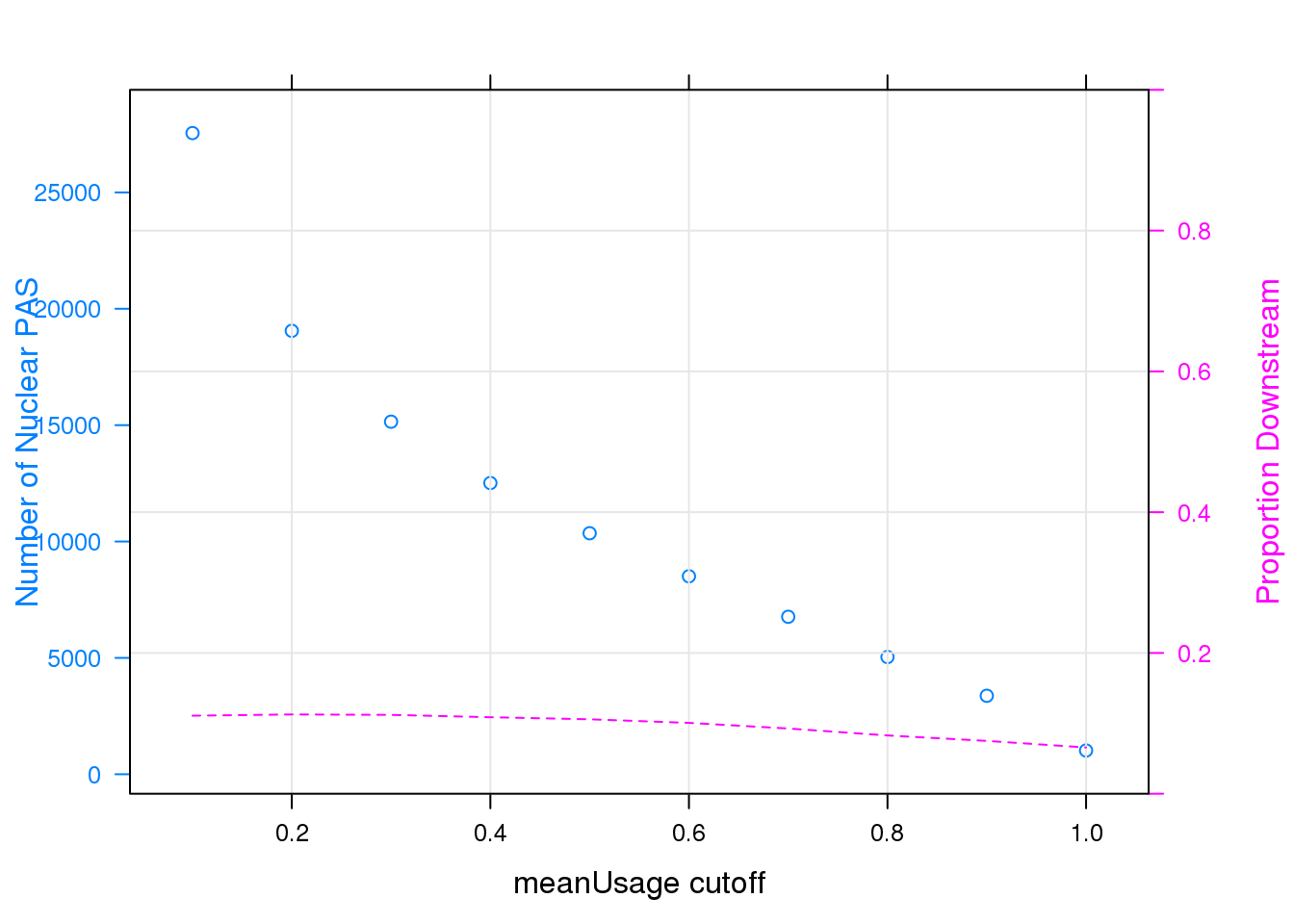
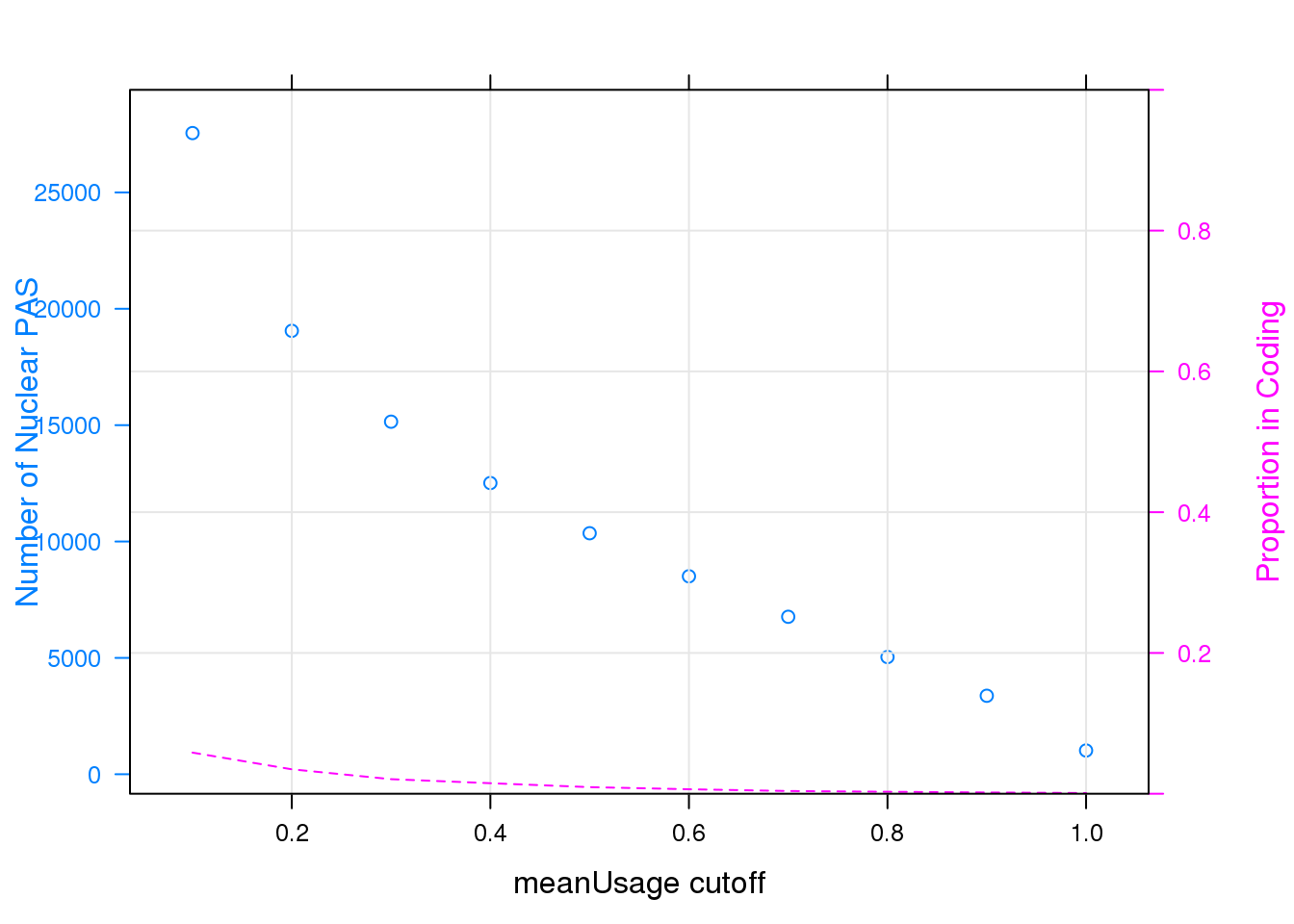
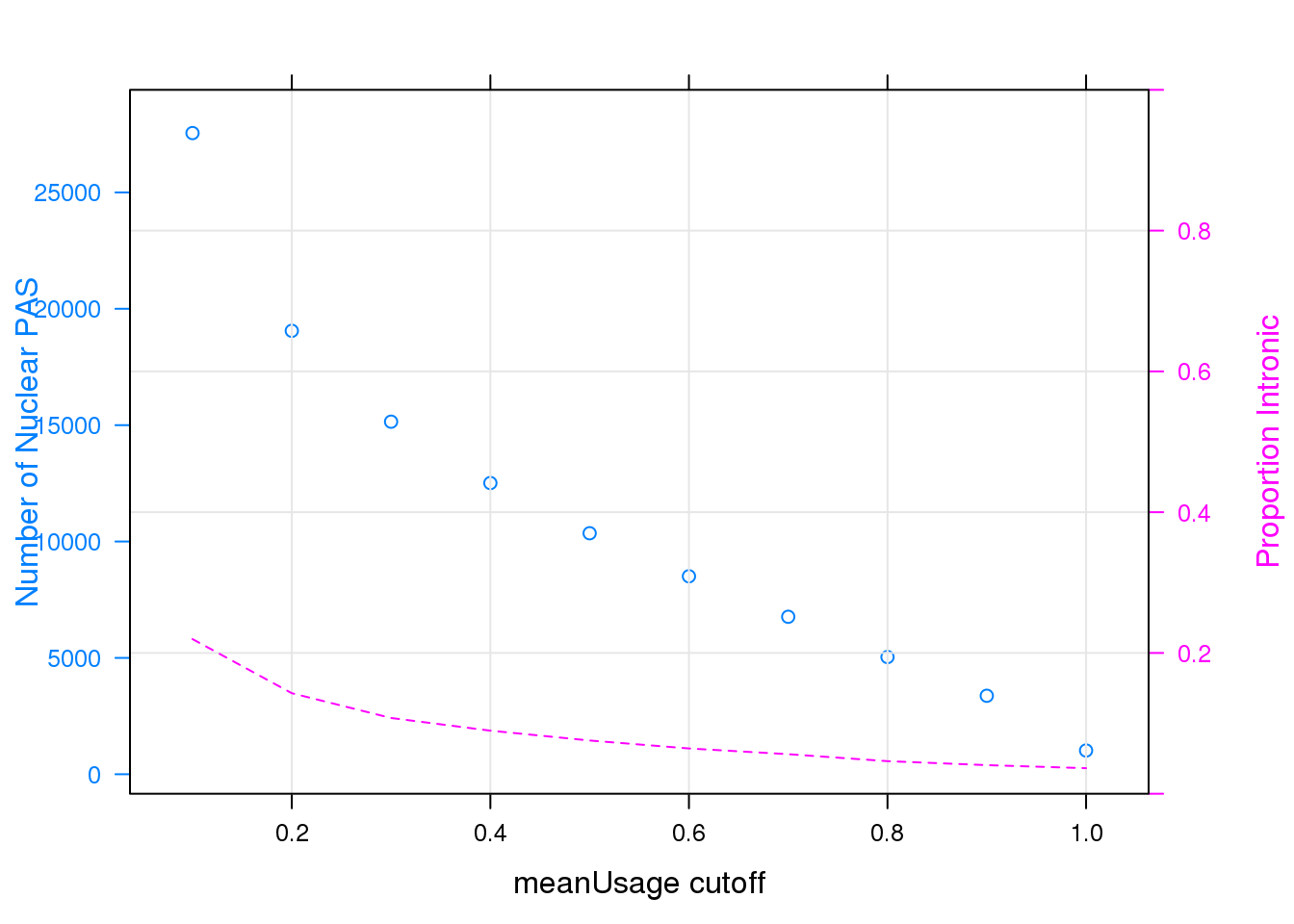
Note that the echo = FALSE parameter was added to the code chunk to prevent printing of the R code that generated the plot.
PDF for the figures:
Remake these with ggplot. I can plot the utr and intron on the same plot. I can make a second plot with the absolute numbers.
Utr3propfixed = Utr3prop %>% dplyr::rename("UTR"=cutoff.numbers)
intronpropfixed= intronprop %>% dplyr::rename("intron"=cutoff.numbers)
utrandIntron=Utr3propfixed %>% inner_join(intronpropfixed,by="xval")
utrandIntron_melt=melt(utrandIntron, id.vars = "xval", variable.name = "location", value.name = "prop")
full=as.data.frame(cbind(cutoff_total, utrandIntron))full_g=full %>% gather("Location", "prop", -cutoff, -xval)
mycolors <- c("data1"="purple")
fixedplotfig1=ggplot(data=full_g, aes(x=xval))+ geom_line(aes(y=cutoff),col="purple",size=3)+ geom_line(aes(x=xval, y=prop*30000 , col=Location),size=3)+ scale_y_continuous(sec.axis = sec_axis( ~./30000 , name="Proportion of PAS")) + labs(y="Number of PAS",title="Intronic PAS are used at low frequencies",x="Usage cutoff")+ scale_color_brewer(palette = "Dark2") +theme(axis.title.y.left = element_text(color = mycolors["data1"]), axis.text.y.left = element_text(color = mycolors["data1"]))fixedplotfig1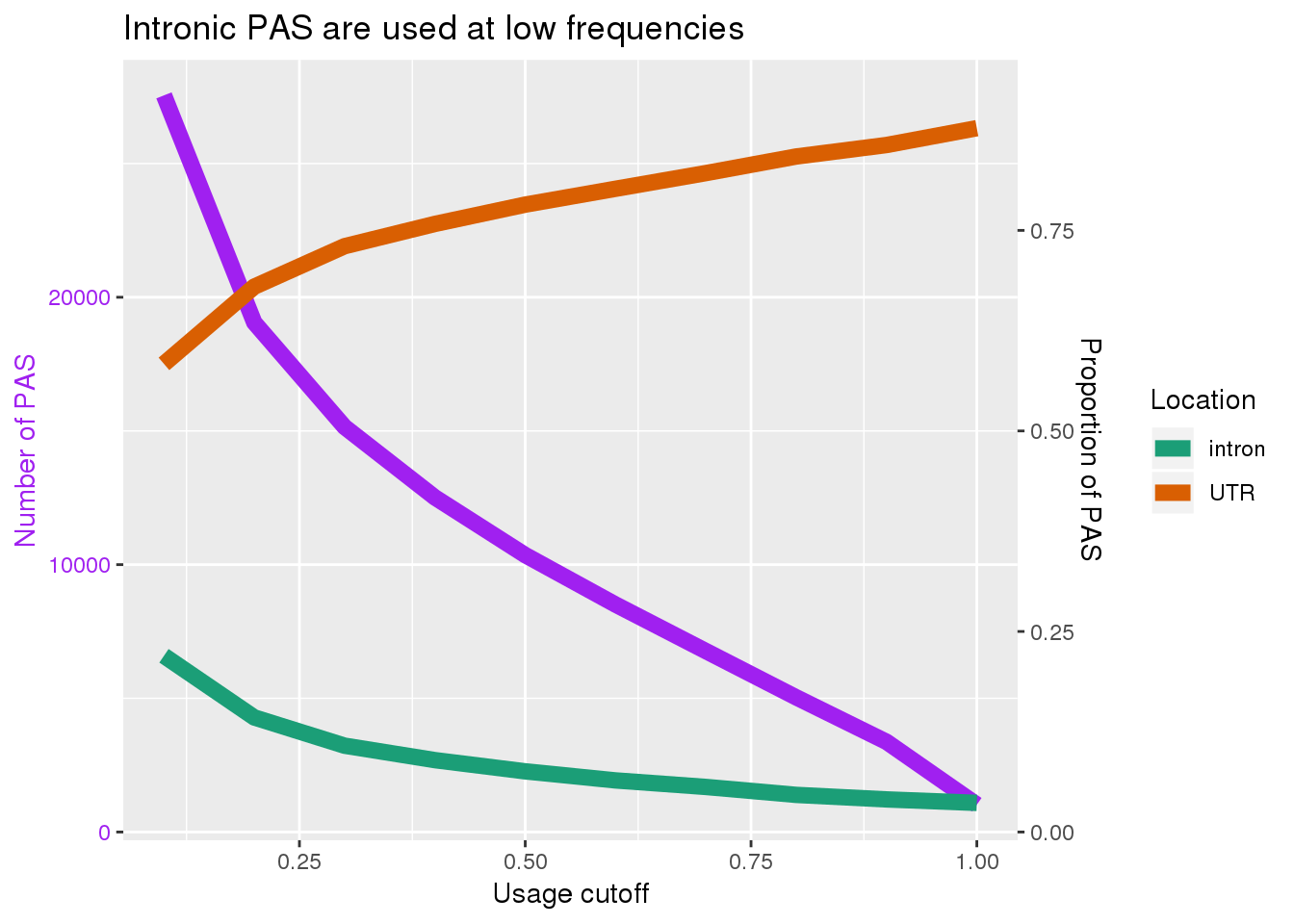
| Version | Author | Date |
|---|---|---|
| 7477dd7 | brimittleman | 2020-02-06 |
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] reshape2_1.4.3 forcats_0.3.0 stringr_1.3.1
[4] purrr_0.3.2 readr_1.3.1 tibble_2.1.1
[7] ggplot2_3.1.1 tidyverse_1.2.1 latticeExtra_0.6-28
[10] RColorBrewer_1.1-2 lattice_0.20-38 tidyr_0.8.3
[13] dplyr_0.8.0.1
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 haven_1.1.2 colorspace_1.3-2 generics_0.0.2
[5] htmltools_0.3.6 yaml_2.2.0 rlang_0.4.0 later_0.7.5
[9] pillar_1.3.1 glue_1.3.0 withr_2.1.2 modelr_0.1.2
[13] readxl_1.1.0 plyr_1.8.4 munsell_0.5.0 gtable_0.2.0
[17] workflowr_1.5.0 cellranger_1.1.0 rvest_0.3.2 evaluate_0.12
[21] labeling_0.3 knitr_1.20 httpuv_1.4.5 broom_0.5.1
[25] Rcpp_1.0.2 promises_1.0.1 backports_1.1.2 scales_1.0.0
[29] jsonlite_1.6 fs_1.3.1 hms_0.4.2 digest_0.6.18
[33] stringi_1.2.4 grid_3.5.1 rprojroot_1.3-2 cli_1.1.0
[37] tools_3.5.1 magrittr_1.5 lazyeval_0.2.1 crayon_1.3.4
[41] whisker_0.3-2 pkgconfig_2.0.2 xml2_1.2.0 lubridate_1.7.4
[45] assertthat_0.2.0 rmarkdown_1.10 httr_1.3.1 rstudioapi_0.10
[49] R6_2.3.0 nlme_3.1-137 git2r_0.26.1 compiler_3.5.1