RNA binding
Briana Mittleman
2/1/2020
Last updated: 2020-02-11
Checks: 7 0
Knit directory: apaQTL/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190411) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/.DS_Store
Ignored: data/ProSeq/
Ignored: output/.DS_Store
Untracked files:
Untracked: .Rprofile
Untracked: ._.DS_Store
Untracked: .gitignore
Untracked: @
Untracked: GEO_brimittleman/
Untracked: _workflowr.yml
Untracked: analysis/._PASdescriptiveplots.Rmd
Untracked: analysis/._cuttoffPercUsage.Rmd
Untracked: analysis/APApeak_Phenotype_GeneLocAnno.Nuclear.5perc.fc.gz.qqnorm.allChrom
Untracked: analysis/APApeak_Phenotype_GeneLocAnno.Total.5perc.fc.gz.qqnorm.allChrom
Untracked: analysis/QTLexampleplots.Rmd
Untracked: analysis/cuttoffPercUsage.Rmd
Untracked: analysis/eQTLoverlap.Rmd
Untracked: analysis/interpret verify bam.Rmd
Untracked: analysis/interpret_verifybam.Rmd
Untracked: analysis/mergeRNA.Rmd
Untracked: analysis/oldstuffNotNeeded.Rmd
Untracked: analysis/remove_badlines.Rmd
Untracked: analysis/totSpecInNuclear.Rmd
Untracked: analysis/totSpecIncludenotTested.Rmd
Untracked: analysis/totalspec.Rmd
Untracked: apaQTL.Rproj
Untracked: checksumsfastq.txt.gz
Untracked: code/.NascentRNAdtPlotFirstintronicPAS.sh.swp
Untracked: code/._ApaQTL_nominalNonnorm.sh
Untracked: code/._BothFracDTPlotGeneRegions.sh
Untracked: code/._BothFracDTPlotGeneRegions_normalized.sh
Untracked: code/._DistPAS2Sig_RandomIntron.py
Untracked: code/._EandPqtl_perm.sh
Untracked: code/._EandPqtls.sh
Untracked: code/._FC_NucintornUpandDown.sh
Untracked: code/._FC_UTR.sh
Untracked: code/._FC_intornUpandDownsteamPAS.sh
Untracked: code/._FC_nascentseq.sh
Untracked: code/._FC_newPeaks_olddata.sh
Untracked: code/._HMMpermuteTotal.py
Untracked: code/._HmmPermute.py
Untracked: code/._IntronicPASDT.sh
Untracked: code/._LC_samplegroups.py
Untracked: code/._LD_qtl.sh
Untracked: code/._LD_snpsproxy.sh
Untracked: code/._MapAllRBP.sh
Untracked: code/._NascentRNAdtPlot.sh
Untracked: code/._NascentRNAdtPlot3UTRPAS.sh
Untracked: code/._NascentRNAdtPlotExcludeFirstintronicPAS.sh
Untracked: code/._NascentRNAdtPlotNucPAS.sh
Untracked: code/._NascentRNAdtPlotTotPAS.sh
Untracked: code/._NascentRNAdtPlotintronicPAS.sh
Untracked: code/._NascnetRNAdtPlotPAS.sh
Untracked: code/._NetSeq_fourthintronDT.sh
Untracked: code/._NomResfromPASSNP.py
Untracked: code/._NuclearPAS_5per.bed.py
Untracked: code/._NuclearandRNA5samp_dtplots.sh
Untracked: code/._PTTfacetboxplots.R
Untracked: code/._PrematureQTLNominal.sh
Untracked: code/._PrematureQTLPermuted.sh
Untracked: code/._QTL2bed.py
Untracked: code/._QTL2bed_withstrand.py
Untracked: code/._RBPdisrupt.sh
Untracked: code/._RNAbam2bw.sh
Untracked: code/._RNAseqDTplot.sh
Untracked: code/._Rplots.pdf
Untracked: code/._RunRes2PAS.sh
Untracked: code/._SAF215upbed.py
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilefiltPAS
Untracked: code/._TESplots100bp.sh
Untracked: code/._TESplots150bp.sh
Untracked: code/._TESplots200bp.sh
Untracked: code/._TotalPAS_5perc.bed.py
Untracked: code/._Untitled
Untracked: code/._ZipandTabPheno.sh
Untracked: code/._aAPAqtl_nominal39ind.sh
Untracked: code/._allNucSpecQTLine.py
Untracked: code/._allNucSpecfromNonNorm.py
Untracked: code/._annotatePacBioPASregion.sh
Untracked: code/._annotatedPAS2bed.py
Untracked: code/._apaInPandE.py
Untracked: code/._apaQTLCorrectPvalMakeQQ.R
Untracked: code/._apaQTLCorrectpval_6or7a.R
Untracked: code/._apaQTL_Nominal.sh
Untracked: code/._apaQTL_nominalInclusive.sh
Untracked: code/._apaQTL_nominalv67.sh
Untracked: code/._apaQTL_permuted.sh
Untracked: code/._apaQTL_permuted_test6A7A.sh
Untracked: code/._apainRibo.py
Untracked: code/._assignNucIntonpeak2intronlocs.sh
Untracked: code/._assignTotIntronpeak2intronlocs.sh
Untracked: code/._bam2BW_5primemost.sh
Untracked: code/._bed2saf.py
Untracked: code/._bothFracDTplot1stintron.sh
Untracked: code/._bothFracDTplot4thintron.sh
Untracked: code/._bothFrac_FC.sh
Untracked: code/._callPeaksYL.py
Untracked: code/._changeRibonomQTLres2genename.py
Untracked: code/._changenomQTLres2geneName.py
Untracked: code/._chooseAnno2PAS_pacbio.py
Untracked: code/._chooseAnno2SAF.py
Untracked: code/._chooseSignalSite
Untracked: code/._chooseSignalSite.py
Untracked: code/._closestannotated.sh
Untracked: code/._closestannotated_byfrac.sh
Untracked: code/._cluster.json
Untracked: code/._clusterPAS.json
Untracked: code/._clusterfiltPAS.json
Untracked: code/._codingdms2bed.py
Untracked: code/._config.yaml
Untracked: code/._config2.yaml
Untracked: code/._configOLD.yaml
Untracked: code/._convertNominal2SNPLOC.py
Untracked: code/._convertNominal2SNPloc2Versions.py
Untracked: code/._convertNumeric.py
Untracked: code/._correctNomeqtl.R
Untracked: code/._createPlinkSampfile.py
Untracked: code/._dag.pdf
Untracked: code/._eQTL_switch2snploc.py
Untracked: code/._eQTLgenestestedapa.py
Untracked: code/._encodeRNADTplots.sh
Untracked: code/._extactPAS100meanphyloP.py
Untracked: code/._extractGenotypes.py
Untracked: code/._extractPACmeanPhyloP.py
Untracked: code/._extractseqfromqtlfastq.py
Untracked: code/._fc2leafphen.py
Untracked: code/._fc_filteredPAS6and7As.sh
Untracked: code/._fifteenBPupstreamPAS.py
Untracked: code/._fiftyBPupstreamPAS.py
Untracked: code/._filter5perc.R
Untracked: code/._filter5percPheno.py
Untracked: code/._filterLDsnps.py
Untracked: code/._filterMPPAS.py
Untracked: code/._filterMPPAS_15.py
Untracked: code/._filterMPPAS_15_7As.py
Untracked: code/._filterMPPAS_50.py
Untracked: code/._filterSAFforMP.py
Untracked: code/._filterpeaks.py
Untracked: code/._finalPASbed2SAF.py
Untracked: code/._fix4su304corr.py
Untracked: code/._fix4su604corr.py
Untracked: code/._fix4sukalisto.py
Untracked: code/._fixExandUnexeQTL
Untracked: code/._fixExandUnexeQTL.py
Untracked: code/._fixFChead.py
Untracked: code/._fixFChead_bothfrac.py
Untracked: code/._fixFChead_short.py
Untracked: code/._fixGWAS4Munge.py
Untracked: code/._fixH3k12ac.py
Untracked: code/._fixPASregionSNPs.py
Untracked: code/._fixRNAhead4corr.py
Untracked: code/._fixRNAkalisto.py
Untracked: code/._fix_randomIntron.py
Untracked: code/._fixgroupedtranscript.py
Untracked: code/._fixhead_netseqfc.py
Untracked: code/._getAPAfromanyeQTL.py
Untracked: code/._getApapval4eqtl.py
Untracked: code/._getApapval4eqtl_unexp.py
Untracked: code/._getApapval4eqtl_version67.py
Untracked: code/._getDownstreamIntronNuclear.py
Untracked: code/._getIntronDownstreamPAS.py
Untracked: code/._getIntronUpstreamPAS.py
Untracked: code/._getQTLalleles.py
Untracked: code/._getQTLfastq.sh
Untracked: code/._getUpstreamIntronNuclear.py
Untracked: code/._grouptranscripts.py
Untracked: code/._intersectVCFandupPAS.sh
Untracked: code/._keep5perMAF.py
Untracked: code/._keepSNP_vcf.sh
Untracked: code/._make5percPeakbed.py
Untracked: code/._makeFileID.py
Untracked: code/._makePheno.py
Untracked: code/._makeSAFbothfrac5perc.py
Untracked: code/._makeSNP2rsidfile.py
Untracked: code/._makeeQTLempirical_unexp.py
Untracked: code/._makeeQTLempiricaldist.py
Untracked: code/._makegencondeTSSfile.py
Untracked: code/._mapSSsnps2PAS.sh
Untracked: code/._mergRNABam.sh
Untracked: code/._mergeAllBam.sh
Untracked: code/._mergeAnnotations.sh
Untracked: code/._mergeBW_norm.sh
Untracked: code/._mergeBamNascent.sh
Untracked: code/._mergeByFracBam.sh
Untracked: code/._mergePeaks.sh
Untracked: code/._miRNAdisrupt.sh
Untracked: code/._mnase1stintron.sh
Untracked: code/._mnaseDT_fourthintron.sh
Untracked: code/._namePeaks.py
Untracked: code/._netseqDTplot1stIntron.sh
Untracked: code/._netseqFC.sh
Untracked: code/._nucQTLGWAS.py
Untracked: code/._nucSpecQTLineData.py
Untracked: code/._nucSpeceffectsize.py
Untracked: code/._nucspecnucPASine.py
Untracked: code/._pQTLsotherdata.py
Untracked: code/._pacbioDT.sh
Untracked: code/._pacbioIntronicDT.sh
Untracked: code/._parseBestbamid.py
Untracked: code/._parseLDRes.py
Untracked: code/._parseLDresBothPAS.sh
Untracked: code/._peak2PAS.py
Untracked: code/._peakFC.sh
Untracked: code/._pheno2countonly.R
Untracked: code/._phenoQTLfromlist.py
Untracked: code/._processYRIgen.py
Untracked: code/._pttQTLsinapaQTL.py
Untracked: code/._qtlRegionseq.sh
Untracked: code/._qtlsPvalOppFrac.py
Untracked: code/._quantassign2parsedpeak.py
Untracked: code/._removeXfromHmm.py
Untracked: code/._removeloc_pheno.py
Untracked: code/._riboQTL.sh
Untracked: code/._runCorrectNomEqtl.sh
Untracked: code/._runFixGWAS4Munge.sh
Untracked: code/._runHMMpermuteAPAqtls.sh
Untracked: code/._runHMMpermuteeQTLS.sh
Untracked: code/._runMakeEmpiricaleQTL_unexp.sh
Untracked: code/._runMakeeQTLempirical.sh
Untracked: code/._run_bam2bw_all3prime.sh
Untracked: code/._run_bam2bw_extra3.sh
Untracked: code/._run_bestbamid.sj
Untracked: code/._run_dist2sig_randomintron.sh
Untracked: code/._run_filtersnpLD.sh
Untracked: code/._run_getAPAfromeQTL_version6.7.sh
Untracked: code/._run_getApaPval4eqtl.sh
Untracked: code/._run_getapafromeQTL.py
Untracked: code/._run_getapafromeQTL.sh
Untracked: code/._run_getapapval4eqtl_unexp.sh
Untracked: code/._run_leafcutterDiffIso.sh
Untracked: code/._run_prxySNP.sh
Untracked: code/._run_pttfacetboxplot.sh
Untracked: code/._run_sepUsagephen.sh
Untracked: code/._run_sepgenobychrom.sh
Untracked: code/._run_verifybam.sh
Untracked: code/._selectNominalPvalues.py
Untracked: code/._sepUsagePhen.py
Untracked: code/._sepgenobychrom.py
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._sortindexRNAbam.sh
Untracked: code/._specAPAinE.py
Untracked: code/._submit-snakemakePAS.sh
Untracked: code/._submit-snakemakefiltPAS.sh
Untracked: code/._subsetAPAnotEorPgene.py
Untracked: code/._subsetAPAnotEorPgene_2versions.py
Untracked: code/._subsetApanoteGene.py
Untracked: code/._subsetApanoteGene_2versions.py
Untracked: code/._subsetUnexplainedeQTLs.py
Untracked: code/._subsetVCF_SS.sh
Untracked: code/._subsetVCF_noSSregions.sh
Untracked: code/._subsetVCF_upstreamPAS.sh
Untracked: code/._subset_diffisopheno.py
Untracked: code/._subsetpermAPAwithGenelist.py
Untracked: code/._subsetpermAPAwithGenelist_2versions.py
Untracked: code/._subsetvcf_otherreg.sh
Untracked: code/._subsetvcf_permSS.sh
Untracked: code/._subtrachfiveprimeUTR.sh
Untracked: code/._subtractExons.sh
Untracked: code/._subtractfiveprimeUTR.sh
Untracked: code/._tabixSNPS.sh
Untracked: code/._tenBPupstreamPAS.py
Untracked: code/._test.pdf
Untracked: code/._testVerifyBam.sh
Untracked: code/._totSeceffectsize.py
Untracked: code/._twentyBPupstreamPAS.py
Untracked: code/._utrdms2saf.py
Untracked: code/._vcf2bed.py
Untracked: code/._verifyBam18517N.sh
Untracked: code/._verifyBam18517T.sh
Untracked: code/._verifyBam19128N.sh
Untracked: code/._verifyBam19128T.sh
Untracked: code/._wrap_verifybam.sh
Untracked: code/._writePTTexamplecode.py
Untracked: code/._writePTTexamplecode.sh
Untracked: code/.pversion
Untracked: code/.snakemake/
Untracked: code/1
Untracked: code/APAqtl_nominal.err
Untracked: code/APAqtl_nominal.out
Untracked: code/APAqtl_nominal_39.err
Untracked: code/APAqtl_nominal_39.out
Untracked: code/APAqtl_nominal_inclusive.err
Untracked: code/APAqtl_nominal_inclusive.out
Untracked: code/APAqtl_nominal_nonNorm.err
Untracked: code/APAqtl_nominal_nonNorm.out
Untracked: code/APAqtl_nominal_versions67.err
Untracked: code/APAqtl_nominal_versions67.out
Untracked: code/APAqtl_permuted.err
Untracked: code/APAqtl_permuted.out
Untracked: code/APAqtl_permuted_versions67.err
Untracked: code/APAqtl_permuted_versions67.out
Untracked: code/BothFracDTPlot1stintron.err
Untracked: code/BothFracDTPlot1stintron.out
Untracked: code/BothFracDTPlot4stintron.err
Untracked: code/BothFracDTPlot4stintron.out
Untracked: code/BothFracDTPlotGeneRegions.err
Untracked: code/BothFracDTPlotGeneRegions.out
Untracked: code/BothFracDTPlotGeneRegions_norm.err
Untracked: code/BothFracDTPlotGeneRegions_norm.out
Untracked: code/DistPAS2Sig_RandomIntron.py
Untracked: code/EandPqtl.err
Untracked: code/EandPqtl.out
Untracked: code/EncodeRNADTPlotGeneRegions.err
Untracked: code/EncodeRNADTPlotGeneRegions.out
Untracked: code/FC_NucintronPASupandDown.err
Untracked: code/FC_NucintronPASupandDown.out
Untracked: code/FC_UTR.err
Untracked: code/FC_UTR.out
Untracked: code/FC_intronPASupandDown.err
Untracked: code/FC_intronPASupandDown.out
Untracked: code/FC_nascent.err
Untracked: code/FC_nascentout
Untracked: code/FC_newPAS_olddata.err
Untracked: code/FC_newPAS_olddata.out
Untracked: code/HmmPermute.p
Untracked: code/IntronicPASDT.err
Untracked: code/IntronicPASDT.out
Untracked: code/LD_vcftools.hap.out
Untracked: code/MapAllRBP.sh
Untracked: code/MapRBP.err
Untracked: code/MapRBP.out
Untracked: code/NascentDTPlotGeneRegions.err
Untracked: code/NascentDTPlotGeneRegions.out
Untracked: code/NascentDTPlotPAS.err
Untracked: code/NascentDTPlotPAS.out
Untracked: code/NascentDTPlotPAS_3utr.err
Untracked: code/NascentDTPlotPAS_3utr.out
Untracked: code/NascentDTPlotPAS_firstintron.err
Untracked: code/NascentDTPlotPAS_firstintron.out
Untracked: code/NascentDTPlotPAS_intron.err
Untracked: code/NascentDTPlotPAS_intron.out
Untracked: code/NascentDTPlotPAS_nuc.err
Untracked: code/NascentDTPlotPAS_nuc.out
Untracked: code/NascentDTPlotPAS_tot.err
Untracked: code/NascentDTPlotPAS_tot.out
Untracked: code/Nuclear_example.err
Untracked: code/Nuclear_example.out
Untracked: code/NuclearandRNA5samp_dtplots.sh
Untracked: code/NuclearandRNAFracDTPlotGeneRegions.err
Untracked: code/NuclearandRNAFracDTPlotGeneRegions.out
Untracked: code/PACbioDT.err
Untracked: code/PACbioDT.out
Untracked: code/PACbioDTitronic.err
Untracked: code/PACbioDTitronic.out
Untracked: code/Prematureqtl_nominal.err
Untracked: code/Prematureqtl_nominal.out
Untracked: code/Prematureqtl_permuted.err
Untracked: code/Prematureqtl_permuted.out
Untracked: code/RBPdisrupt.err
Untracked: code/RBPdisrupt.out
Untracked: code/RBPdisrupt.sh
Untracked: code/README.md
Untracked: code/RNABam2BW.err
Untracked: code/RNABam2BW.out
Untracked: code/RNAseqDTPlotGeneRegions.err
Untracked: code/RNAseqDTPlotGeneRegions.out
Untracked: code/Rplots.pdf
Untracked: code/TESplots100bp.err
Untracked: code/TESplots100bp.out
Untracked: code/TESplots150bp.err
Untracked: code/TESplots150bp.out
Untracked: code/TESplots200bp.err
Untracked: code/TESplots200bp.out
Untracked: code/Total_example.err
Untracked: code/Total_example.out
Untracked: code/Untitled
Untracked: code/YRI_LCL.vcf.gz
Untracked: code/YRI_LCL_chr1.vcf.gz.log
Untracked: code/YRI_LCL_chr1.vcf.gz.recode.vcf
Untracked: code/annotatedPASregion.err
Untracked: code/annotatedPASregion.out
Untracked: code/apaQTL_nominalInclusive.sh
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assigntotPeak2Intronicregion.err
Untracked: code/assigntotPeak2Intronicregion.out
Untracked: code/bam2bw.err
Untracked: code/bam2bw.out
Untracked: code/bam2bw_5primemost.err
Untracked: code/bam2bw_5primemost.out
Untracked: code/binary_fileset.log
Untracked: code/bothFrac_FC.err
Untracked: code/bothFrac_FC.out
Untracked: code/callSHscripts.txt
Untracked: code/closestannotated.err
Untracked: code/closestannotated.out
Untracked: code/closestannotatedbyfrac.err
Untracked: code/closestannotatedbyfrac.out
Untracked: code/dag.pdf
Untracked: code/dagPAS.pdf
Untracked: code/dagfiltPAS.pdf
Untracked: code/extactPAS100meanphyloP.py
Untracked: code/extractPACmeanPhyloP.py
Untracked: code/fixExandUnexeQTL
Untracked: code/fixGWAS4Munge.py
Untracked: code/fix_randomIntron.py
Untracked: code/fixmunge
Untracked: code/genotypesYRI.gen.proc.keep.vcf.log
Untracked: code/genotypesYRI.gen.proc.keep.vcf.recode.vcf
Untracked: code/getseq100up.err
Untracked: code/getseq100up.out
Untracked: code/grouptranscripts.err
Untracked: code/grouptranscripts.out
Untracked: code/intersectPAS_ssSNPS.err
Untracked: code/intersectPAS_ssSNPS.out
Untracked: code/intersectVCFPAS.err
Untracked: code/intersectVCFPAS.out
Untracked: code/log/
Untracked: code/logs/
Untracked: code/merge53PRIMEbam.err
Untracked: code/merge53PRIMEbam.out
Untracked: code/merge53RNAbam.err
Untracked: code/merge53prime.sh
Untracked: code/merge5RNABam.err
Untracked: code/merge5RNABam.out
Untracked: code/merge5RNAbam.out
Untracked: code/merge5RNAbam.sh
Untracked: code/mergeAnno.err
Untracked: code/mergeAnno.out
Untracked: code/mergeBWnorm.err
Untracked: code/mergeBWnorm.out
Untracked: code/mergeBamNacent.err
Untracked: code/mergeBamNacent.out
Untracked: code/mergeRNAbam.err
Untracked: code/mergeRNAbam.out
Untracked: code/miRNAdisrupt.err
Untracked: code/miRNAdisrupt.out
Untracked: code/miRNAdisrupt.sh
Untracked: code/mnaseDTPlot1stintron.err
Untracked: code/mnaseDTPlot1stintron.out
Untracked: code/mnaseDTPlot4thintron.err
Untracked: code/mnaseDTPlot4thintron.out
Untracked: code/netDTPlot4thintron.out
Untracked: code/netseqFC.err
Untracked: code/netseqFC.out
Untracked: code/neyDTPlot4thintron.err
Untracked: code/nucspecinE.py
Untracked: code/parseLDRes.py
Untracked: code/parseLDres.err
Untracked: code/parseLDres.out
Untracked: code/parseLDresBothPAS.sh
Untracked: code/plink.log
Untracked: code/prxySNP.err
Untracked: code/prxySNP.out
Untracked: code/pttFacetBoxplots.err
Untracked: code/pttFacetBoxplots.out
Untracked: code/qtlFacetBoxplots.err
Untracked: code/qtlFacetBoxplots.out
Untracked: code/rLD_vcftools.hap.err
Untracked: code/riboqtl.err
Untracked: code/riboqtl.out
Untracked: code/runBestBamID.err
Untracked: code/runCorrectNomeqtl.err
Untracked: code/runCorrectNomeqtl.out
Untracked: code/runFilterLD.err
Untracked: code/runFilterLD.out
Untracked: code/runFixGWAS4Munge.sh
Untracked: code/runHMMpermute.err
Untracked: code/runHMMpermute.out
Untracked: code/runHMMpermuteeQTLs.err
Untracked: code/runHMMpermuteeQTLs.out
Untracked: code/runMakeEmpiricaleQTLs.err
Untracked: code/runMakeEmpiricaleQTLs.out
Untracked: code/runMakeEmpiricaleQTLsunex.err
Untracked: code/runMakeEmpiricaleQTLsunex.out
Untracked: code/run_DistPAS2Sig.err
Untracked: code/run_DistPAS2Sig.out
Untracked: code/run_DistPAS2Sig_intron.err
Untracked: code/run_DistPAS2Sig_intron.out
Untracked: code/run_bam2bw.err
Untracked: code/run_bam2bw.out
Untracked: code/run_bam2bwexta.err
Untracked: code/run_bam2bwexta.out
Untracked: code/run_dist2sig_randomintron.sh
Untracked: code/run_getAPAfromanyeQTL.err
Untracked: code/run_getAPAfromanyeQTL.out
Untracked: code/run_getApaPval4eQTLs.err
Untracked: code/run_getApaPval4eQTLs.out
Untracked: code/run_getApaPval4eQTLsunexplained.err
Untracked: code/run_getApaPval4eQTLsunexplained.out
Untracked: code/run_leafcutter_ds.err
Untracked: code/run_leafcutter_ds.out
Untracked: code/run_sepgenobychrom.err
Untracked: code/run_sepgenobychrom.out
Untracked: code/run_sepusage.err
Untracked: code/run_sepusage.out
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/run_verifybam128N.err
Untracked: code/run_verifybam128N.out
Untracked: code/run_verifybam128T.err
Untracked: code/run_verifybam128T.out
Untracked: code/run_verifybam517N.err
Untracked: code/run_verifybam517N.out
Untracked: code/run_verifybam517T.err
Untracked: code/run_verifybam517T.out
Untracked: code/runprxySNP.err
Untracked: code/runprxySNP.out
Untracked: code/runres2pas.err
Untracked: code/runres2pas.out
Untracked: code/scripts/
Untracked: code/scripts_PAS_500_Lymph/
Untracked: code/seqQTLfastq.err
Untracked: code/seqQTLfastq.out
Untracked: code/seqQTLregion.err
Untracked: code/seqQTLregion.out
Untracked: code/snakePASlog.out
Untracked: code/snakefiltPASlog.out
Untracked: code/sortindexRNABam.err
Untracked: code/sortindexRNABam.out
Untracked: code/specAPAinE.py
Untracked: code/subsetvcf_SS.err
Untracked: code/subsetvcf_SS.out
Untracked: code/subsetvcf_noSS.err
Untracked: code/subsetvcf_noSS.out
Untracked: code/subsetvcf_pas.err
Untracked: code/subsetvcf_pas.out
Untracked: code/subsetvcf_perm.err
Untracked: code/subsetvcf_perm.out
Untracked: code/subsetvcf_rand.err
Untracked: code/subsetvcf_rand.out
Untracked: code/subtract5UTR.err
Untracked: code/subtract5UTR.out
Untracked: code/subtractExons.err
Untracked: code/subtractExons.out
Untracked: code/tabixSNPs.err
Untracked: code/tabixSNPs.out
Untracked: code/test.pdf
Untracked: code/testFix.txt
Untracked: code/test_verifybam.err
Untracked: code/test_verifybam.out
Untracked: code/vcf_keepsnps.err
Untracked: code/vcf_keepsnps.out
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/zipandtabPhen.err
Untracked: code/zipandtabPhen.out
Untracked: data/._.DS_Store
Untracked: data/._MetaDataSequencing.txt
Untracked: data/AnnotatedPAS/
Untracked: data/ApaByEgene/
Untracked: data/ApaByPgene/
Untracked: data/BadLines/
Untracked: data/BaseComp/
Untracked: data/Battle_pQTL/
Untracked: data/CheckSums/
Untracked: data/CompareOldandNew/
Untracked: data/DTmatrix/
Untracked: data/DiffIso/
Untracked: data/EncodeRNA/
Untracked: data/ExampleQTLPlots/
Untracked: data/ExampleQTLPlots_update/
Untracked: data/ExpressionIndependentapaQTLs.txt
Untracked: data/FiveMergedBW/
Untracked: data/FiveMergedBam/
Untracked: data/FlaggedPAS/
Untracked: data/GWAS_overlap/
Untracked: data/GeuvadisRNA/
Untracked: data/HMMqtls/
Untracked: data/LDSR_annotations/
Untracked: data/Li_eQTLs/
Untracked: data/NMD/
Untracked: data/NascentRNA/
Untracked: data/NucSpeceQTLeffect/
Untracked: data/PAS/
Untracked: data/PAS_postFlag/
Untracked: data/PolyA_DB/
Untracked: data/PreTerm_pheno/
Untracked: data/PrematureQTLNominal/
Untracked: data/PrematureQTLPermuted/
Untracked: data/QTLGenotypes/
Untracked: data/QTLoverlap/
Untracked: data/QTLoverlap_inclusive/
Untracked: data/QTLoverlap_nonNorm/
Untracked: data/README.md
Untracked: data/RNAseq/
Untracked: data/Reads2UTR/
Untracked: data/SNPinSS/
Untracked: data/SignalSiteFiles/
Untracked: data/TF_motifdisruption/
Untracked: data/TSS/
Untracked: data/ThirtyNineIndQtl_nominal/
Untracked: data/Version15bp6As/
Untracked: data/Version15bp7As/
Untracked: data/apaQTLNominal/
Untracked: data/apaQTLNominal_4pc/
Untracked: data/apaQTLNominal_inclusive/
Untracked: data/apaQTLPermuted/
Untracked: data/apaQTLPermuted_4pc/
Untracked: data/apaQTLs/
Untracked: data/assignedPeaks/
Untracked: data/assignedPeaks_15Up/
Untracked: data/bam/
Untracked: data/bam_clean/
Untracked: data/bam_waspfilt/
Untracked: data/bed_10up/
Untracked: data/bed_clean/
Untracked: data/bed_clean_sort/
Untracked: data/bed_waspfilter/
Untracked: data/bedsort_waspfilter/
Untracked: data/bothFrac_FC/
Untracked: data/bw/
Untracked: data/bw_norm/
Untracked: data/eCLip/
Untracked: data/eQTLs/
Untracked: data/exampleQTLs/
Untracked: data/exosome/
Untracked: data/fastq/
Untracked: data/filterPeaks/
Untracked: data/fourSU/
Untracked: data/h3k27ac/
Untracked: data/highdiffsiggenes.txt
Untracked: data/inclusivePeaks/
Untracked: data/inclusivePeaks_FC/
Untracked: data/intronRNAratio/
Untracked: data/intron_analysis/
Untracked: data/locusZoom/
Untracked: data/mergedBG/
Untracked: data/mergedBW_byfrac/
Untracked: data/mergedBW_norm/
Untracked: data/mergedBam/
Untracked: data/mergedbyFracBam/
Untracked: data/miRNAbinding/
Untracked: data/molPhenos/
Untracked: data/molQTLs/
Untracked: data/motifdistrupt/
Untracked: data/nPAS/
Untracked: data/netseq/
Untracked: data/nonNorm_pheno/
Untracked: data/nuc_10up/
Untracked: data/nuc_10upclean/
Untracked: data/oldPASfiles/
Untracked: data/overlapeQTL_try2/
Untracked: data/overlapeQTLs/
Untracked: data/pQTLoverlap/
Untracked: data/pacbio/
Untracked: data/peakCoverage/
Untracked: data/peaks_5perc/
Untracked: data/phenotype/
Untracked: data/phenotype_5perc/
Untracked: data/phenotype_inclusivePAS/
Untracked: data/phylop/
Untracked: data/pttQTL/
Untracked: data/pttQTLplots/
Untracked: data/sigDiffGenes.txt
Untracked: data/sort/
Untracked: data/sort_clean/
Untracked: data/sort_waspfilter/
Untracked: data/twoMech/
Untracked: data/vareQTLvarAPAqtl/
Untracked: data/verifyBAM/
Untracked: data/verifyBAM_full/
Untracked: nohup.out
Untracked: output/._.DS_Store
Untracked: output/._AverageDiffHeatmap.Nuclear.png
Untracked: output/._AverageDiffHeatmap.Total.png
Untracked: output/._GeneswithAPApotential.png
Untracked: output/._GeneswithAPApotentialAllPAS.png
Untracked: output/._PASlocation.png
Untracked: output/._SignalSitePlot.png
Untracked: output/._meanCorrelationPhenotypes.svg
Untracked: output/._qqplot_Nuclear_APAperm.png
Untracked: output/._qqplot_Nuclear_APAperm_4pc.png
Untracked: output/._qqplot_Total_APAperm.png
Untracked: output/._qqplot_Total_APAperm_4pc.png
Untracked: output/AverageDiffHeatmap.Nuclear.png
Untracked: output/AverageDiffHeatmap.Total.png
Untracked: output/GeneswithAPApotential.png
Untracked: output/GeneswithAPApotentialAllPAS.png
Untracked: output/PASlocation.png
Untracked: output/SignalSitePlot.png
Untracked: output/SignalSitePlotbyLoc.png
Untracked: output/dtPlots/
Untracked: output/fastqc/
Untracked: output/meanCorrelationPhenotypes.svg
Untracked: output/newnuc.png
Untracked: output/newtot.png
Untracked: output/oldnuc.png
Untracked: output/oldtot.png
Untracked: output/qqplot_Nuclear_APAperm.png
Untracked: output/qqplot_Nuclear_APAperm_4pc.png
Untracked: output/qqplot_Total_APAperm.png
Untracked: output/qqplot_Total_APAperm_4pc.png
Untracked: run_verifybam517N.err
Untracked: run_verifybam517N.out
Unstaged changes:
Modified: analysis/ExploreNpas.Rmd
Modified: analysis/NuclearSpecIncludeNotTested.Rmd
Modified: analysis/PASdescriptiveplots.Rmd
Modified: analysis/Readdistagainstfeatures.Rmd
Modified: analysis/TSS.Rmd
Modified: analysis/decayAndStability.Rmd
Modified: analysis/miRNAdisrupt.Rmd
Modified: analysis/nascenttranscription.Rmd
Modified: analysis/nucSpecinEQTLs.Rmd
Modified: analysis/overlapapaqtlsandeqtls.Rmd
Modified: analysis/pQTLexampleplot.Rmd
Modified: analysis/propeQTLs_explained.Rmd
Modified: analysis/version15bpfilter.Rmd
Modified: code/DistPAS2Sig.py
Modified: code/Script4NuclearQTLexamples.sh
Modified: code/Script4TotalQTLexamples.sh
Modified: code/apaQTLsnake.err
Modified: code/run_qtlFacetBoxplots.sh
Deleted: code/test.txt
Deleted: reads_graphs.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 3414736 | brimittleman | 2020-02-11 | add TvN analysis |
| html | f006ad3 | brimittleman | 2020-02-11 | Build site. |
| Rmd | 13c1e8f | brimittleman | 2020-02-11 | add examples for binding disruption |
| html | 1c3d3e7 | brimittleman | 2020-02-10 | Build site. |
| Rmd | 9ac4e5f | brimittleman | 2020-02-10 | add upf1 |
| html | bd6d99c | brimittleman | 2020-02-02 | Build site. |
| Rmd | fda9908 | brimittleman | 2020-02-02 | add RBP res |
library(workflowr)This is workflowr version 1.6.0
Run ?workflowr for help getting startedlibrary(tidyverse)── Attaching packages ───────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ──────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()I will use eClip data from encode to study RNA binding. I will use the results from K562 cells. They do not have data for LCLs.
Downloading the bed files for 25 different proteins.
mkdir ../data/eCLip/I will search in all of the gene UTRs for each of these. I will see if there is more likely to be an overlap in genes with APA.
I need to cut the CHR from each file.
for i in $(ls ../data/eCLip/*.bed)
do
name=$(echo ${i} | cut -f 4 -d '/' | cut -f 1 -d '.')
sed 's/^chr//' $i > ../data/eCLip/${name}.noCHR.bed
doneRun overlap for all othese with bedtools. I will make a bedfile with the longest UTR annoation for each gene.
UTR=read.table("../../genome_anotation_data/RefSeq_annotations/ncbiRefSeq_UTR3.sort.bed",col.names = c('chr','start','end','utr','gene', 'score','strand'),stringsAsFactors = F) %>%
mutate(UTRlength=end-start) %>%
group_by(gene)%>%
arrange(desc(UTRlength)) %>%
filter(row_number() == 1L) %>%
select(chr, start,end, gene, score, strand)
write.table(UTR, "../data/eCLip/UTRregions.bed", row.names = F, col.names = F, quote = F,sep="\t")sort -k1,1 -k2,2n ../data/eCLip/UTRregions.bed > ../data/eCLip/UTRregions.sort.bedMerge the regions with the name being the name of the RNA.
cat ../data/eCLip/*.noCHR.bed > ../data/eCLip/ALLRBP.noCHR.bed
sort -k1,1 -k2,2n ../data/eCLip/ALLRBP.noCHR.bed | cut -f 1-6 > ../data/eCLip/ALLRBP.noCHR.sort.bedcat: ../data/eCLip/ALLRBP.noCHR.bed: input file is output fileNow I can map.Print the distinct RBP in each UTR.
bedtools map -a ../data/eCLip/UTRregions.sort.bed -b ../data/eCLip/ALLRBP.noCHR.sort.bed -c 4 -o distinct -s > ../data/eCLip/AllUTRsMappedallRBP.txtI will also run this all seperatly for downstream analysis:
sbatch MapAllRBP.shI will compare genes with and without QTLs.
QTL_genes=read.table("../data/apaQTLs/NuclearapaQTLGenes.txt",col.names = "gene",stringsAsFactors = F)
QTLTested_genes=read.table("../data/apaQTLs/TestedNuclearapaQTLGenes.txt",col.names = "gene",stringsAsFactors = F) %>% mutate(QTL=ifelse(gene %in% QTL_genes$gene, "Yes","No"))PHF6=read.table("../data/eCLip/UTRregions_ENCFF016IHL_PHF6.txt",header=F, col.names = c('chr','start','end','gene','score','strand','RBP'),stringsAsFactors = F) %>% inner_join(QTLTested_genes, by='gene') %>% mutate(PHF6=ifelse(RBP=="PHF6_K562_rep01", "Yes","No"))x=nrow(PHF6 %>% filter(PHF6=="Yes", QTL=="Yes"))
m= nrow(PHF6 %>% filter(PHF6=="Yes"))
n=nrow(PHF6 %>% filter(PHF6!="Yes"))
k=nrow(PHF6 %>% filter(QTL=="Yes"))
#expected
which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k)))[1] 77#actual:
x[1] 77#pval
phyper(x,m,n,k,lower.tail=F)[1] 0.8531311Test for any RBP:
All=read.table("../data/eCLip/AllUTRsMappedallRBP.txt",header=F, col.names = c('chr','start','end','gene','score','strand','RBP'),stringsAsFactors = F) %>%
inner_join(QTLTested_genes, by='gene') %>%
mutate(HasRBP=ifelse(RBP!=".", "Yes","No"))x=nrow(All %>% filter(HasRBP=="Yes", QTL=="Yes"))
m= nrow(All %>% filter(HasRBP=="Yes"))
n=nrow(All %>% filter(HasRBP!="Yes"))
k=nrow(All %>% filter(QTL=="Yes"))
#expected
which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k)))[1] 423#actual:
x[1] 444#pval
phyper(x,m,n,k,lower.tail=F)[1] 0.0195104This means genes with QTLs are enriched for genes with an identified RBP in the its UTR.
Let’s look at this by the location of QTL.
QTL_intron=read.table("../data/apaQTLs/Nuclear_apaQTLs4pc_5fdr.bed",stringsAsFactors = F,header = T) %>%
separate(name, into=c("gene", "PAS","loc"),sep=":") %>%
filter(loc=="intron")
QTL_UTR=read.table("../data/apaQTLs/Nuclear_apaQTLs4pc_5fdr.bed",stringsAsFactors = F,header = T) %>%
separate(name, into=c("gene", "PAS","loc"),sep=":") %>%
filter(loc=="utr3")
QTLTested_intron=read.table("../data/apaQTLs/TestedNuclearapaQTLGenes.txt",col.names = "gene",stringsAsFactors = F) %>% mutate(QTL=ifelse(gene %in% QTL_intron$gene, "Yes","No"))
QTLTested_utr=read.table("../data/apaQTLs/TestedNuclearapaQTLGenes.txt",col.names = "gene",stringsAsFactors = F) %>% mutate(QTL=ifelse(gene %in% QTL_UTR$gene, "Yes","No"))All_intron=read.table("../data/eCLip/AllUTRsMappedallRBP.txt",header=F, col.names = c('chr','start','end','gene','score','strand','RBP'),stringsAsFactors = F) %>%
inner_join(QTLTested_intron, by='gene') %>%
mutate(HasRBP=ifelse(RBP!=".", "Yes","No"))
x=nrow(All_intron %>% filter(HasRBP=="Yes", QTL=="Yes"))
m= nrow(All_intron %>% filter(HasRBP=="Yes"))
n=nrow(All_intron %>% filter(HasRBP!="Yes"))
k=nrow(All_intron %>% filter(QTL=="Yes"))
#expected
which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k)))[1] 120#actual:
x[1] 120#pval
phyper(x,m,n,k,lower.tail=F)[1] 0.7092743All_UTR=read.table("../data/eCLip/AllUTRsMappedallRBP.txt",header=F, col.names = c('chr','start','end','gene','score','strand','RBP'),stringsAsFactors = F) %>%
inner_join(QTLTested_utr, by='gene') %>%
mutate(HasRBP=ifelse(RBP!=".", "Yes","No"))
x=nrow(All_UTR %>% filter(HasRBP=="Yes", QTL=="Yes"))
m= nrow(All_UTR %>% filter(HasRBP=="Yes"))
n=nrow(All_UTR %>% filter(HasRBP!="Yes"))
k=nrow(All_UTR %>% filter(QTL=="Yes"))
#expected
which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k)))[1] 191#actual:
x[1] 205#pval
phyper(x,m,n,k,lower.tail=F)[1] 0.01927862This is only significant for genes with QTL’s associated with the 3’ UTR.
Try to find which RBP is driving the assocations.
I want a function that will go through each of the RBPs and test for this enrichment association.
for i in $(ls ../data/eCLip/ENCFF*.small.noCHR.bed)
do
name=$(echo ${i} | cut -f 4 -d '/' | cut -f 1 -d '.')
echo $name >> ../data/eCLip/RBPtested.txt
doneRBP_names=read.table("../data/eCLip/RBPtested.txt", col.names = "RBP",stringsAsFactors = F)expected=c()
actual=c()
pval=c()
for (RBP in RBP_names$RBP){
RBPfile=read.table(paste("../data/eCLip/UTRregions_", RBP,".txt", sep=""),header=F, col.names = c('chr','start','end','gene','score','strand','RBP'), stringsAsFactors = F) %>%
inner_join(QTLTested_genes, by='gene') %>%
mutate(HasRBP=ifelse(RBP!=".", "Yes","No"))
x=nrow(RBPfile %>% filter(HasRBP=="Yes", QTL=="Yes"))
m= nrow(RBPfile %>% filter(HasRBP=="Yes"))
n=nrow(RBPfile %>% filter(HasRBP!="Yes"))
k=nrow(RBPfile %>% filter(QTL=="Yes"))
expected=c(expected, which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k))))
actual=c(actual,x)
pval=c(pval,phyper(x,m,n,k,lower.tail=F))
}RBP_names_res= as.data.frame(cbind(RBP=RBP_names$RBP, expected,actual,pval)) %>% separate(RBP, into=c("exp", "protein"),sep="_")
RBP_names_res$pval=as.numeric(as.character(RBP_names_res$pval))
ggplot(RBP_names_res, aes(x=protein, y=-log10(pval),fill=protein))+geom_bar(stat="identity") +theme(legend.position = "none", axis.text.x = element_text(angle = 90)) +labs(title="Enrichment for nuclear apaQTL genes with RBP in UTR",y="-log10(Enrichment pval)") + geom_hline(yintercept = 2)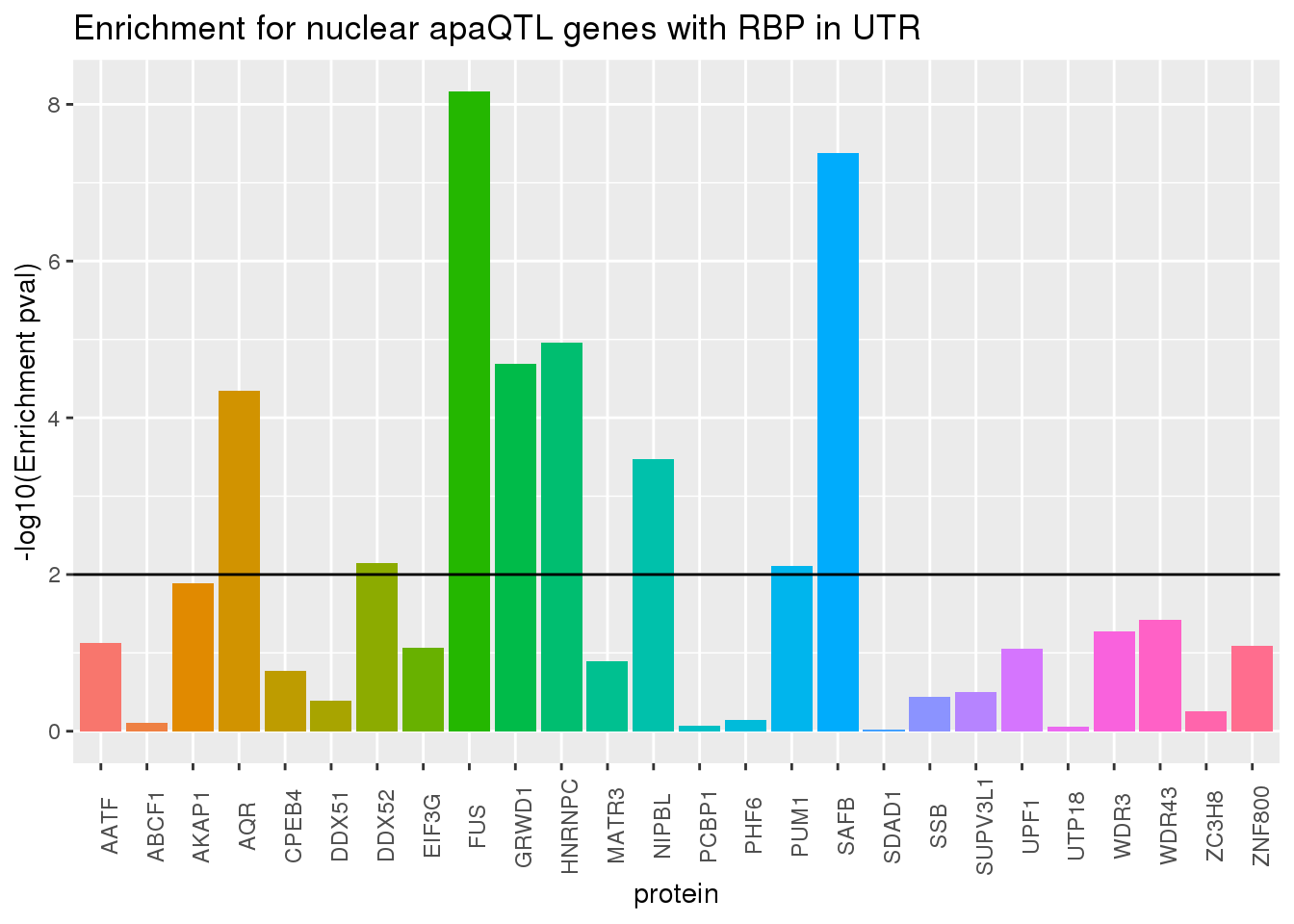
Do this for UTR QTL
expectedUTR=c()
actualUTR=c()
pvalUTR=c()
for (RBP in RBP_names$RBP){
RBPfile=read.table(paste("../data/eCLip/UTRregions_", RBP,".txt", sep=""),header=F, col.names = c('chr','start','end','gene','score','strand','RBP'), stringsAsFactors = F) %>%
inner_join(QTLTested_utr, by='gene') %>%
mutate(HasRBP=ifelse(RBP!=".", "Yes","No"))
x=nrow(RBPfile %>% filter(HasRBP=="Yes", QTL=="Yes"))
m= nrow(RBPfile %>% filter(HasRBP=="Yes"))
n=nrow(RBPfile %>% filter(HasRBP!="Yes"))
k=nrow(RBPfile %>% filter(QTL=="Yes"))
expectedUTR=c(expectedUTR, which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k))))
actualUTR=c(actualUTR,x)
pvalUTR=c(pvalUTR,phyper(x,m,n,k,lower.tail=F))
}RBP_names_resUTR= as.data.frame(cbind(RBP=RBP_names$RBP, expectedUTR,actualUTR,pvalUTR)) %>% separate(RBP, into=c("exp", "protein"),sep="_")
RBP_names_resUTR$pvalUTR=as.numeric(as.character(RBP_names_resUTR$pvalUTR))
ggplot(RBP_names_resUTR, aes(x=protein, y=-log10(pvalUTR),fill=protein))+geom_bar(stat="identity") +theme(legend.position = "none", axis.text.x = element_text(angle = 90)) +labs(title="Enrichment for 3' UTR nuclear apaQTL genes with RBP in UTR",y="-log10(Enrichment pval)") + geom_hline(yintercept = 2)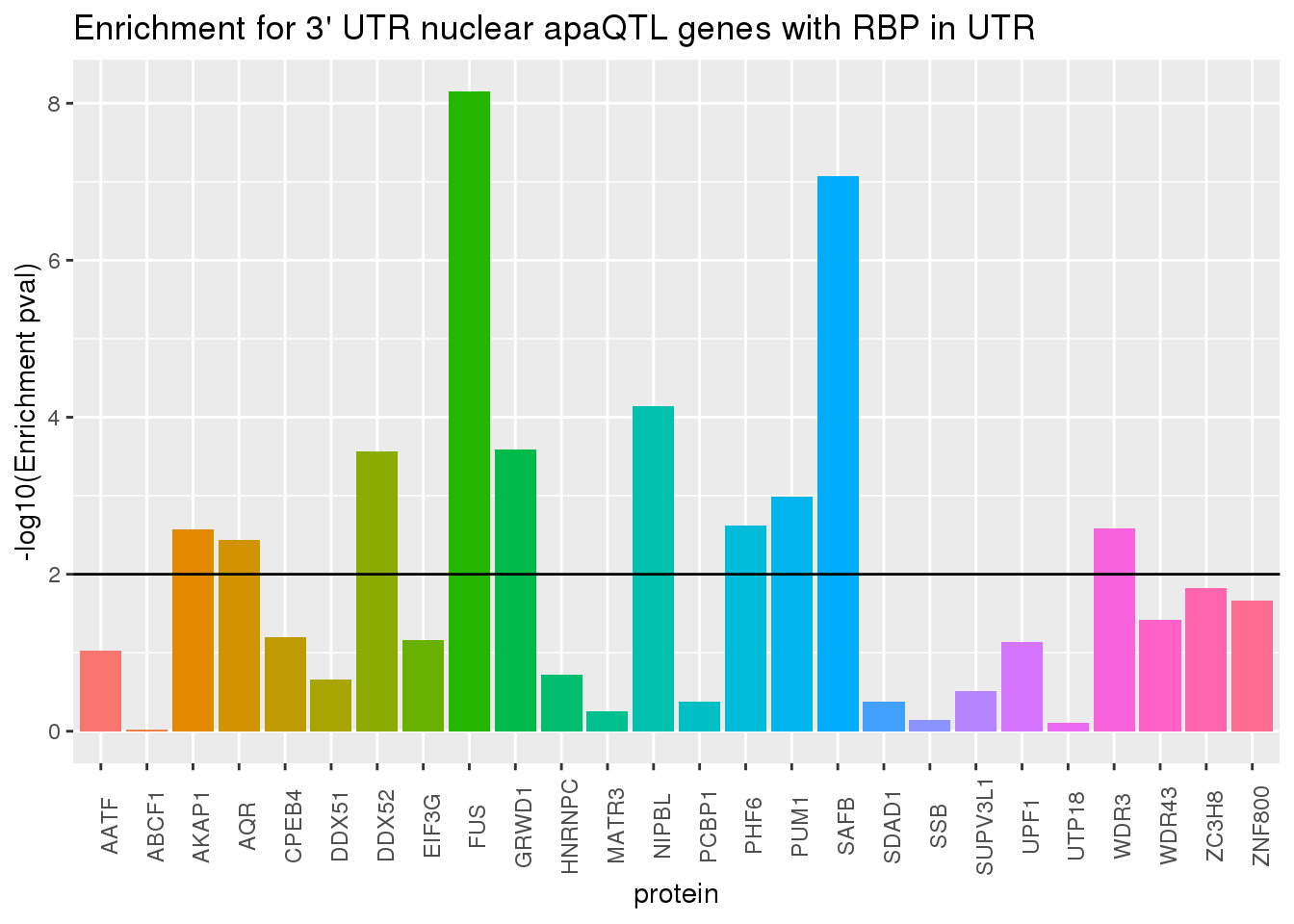
Intronic:
expectedIntron=c()
actualIntron=c()
pvalIntron=c()
for (RBP in RBP_names$RBP){
RBPfile=read.table(paste("../data/eCLip/UTRregions_", RBP,".txt", sep=""),header=F, col.names = c('chr','start','end','gene','score','strand','RBP'), stringsAsFactors = F) %>%
inner_join(QTLTested_intron, by='gene') %>%
mutate(HasRBP=ifelse(RBP!=".", "Yes","No"))
x=nrow(RBPfile %>% filter(HasRBP=="Yes", QTL=="Yes"))
m= nrow(RBPfile %>% filter(HasRBP=="Yes"))
n=nrow(RBPfile %>% filter(HasRBP!="Yes"))
k=nrow(RBPfile %>% filter(QTL=="Yes"))
expectedIntron=c(expectedIntron, which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k))))
actualIntron=c(actualIntron,x)
pvalIntron=c(pvalIntron,phyper(x,m,n,k,lower.tail=F))
}RBP_names_resIntron= as.data.frame(cbind(RBP=RBP_names$RBP, expectedIntron,actualIntron,pvalIntron)) %>% separate(RBP, into=c("exp", "protein"),sep="_")
RBP_names_resIntron$pvalIntron=as.numeric(as.character(RBP_names_resIntron$pvalIntron))
ggplot(RBP_names_resIntron, aes(x=protein, y=-log10(pvalIntron),fill=protein))+geom_bar(stat="identity") +theme(legend.position = "none", axis.text.x = element_text(angle = 90)) +labs(title="Enrichment for Intronic nuclear apaQTL genes with RBP in UTR",y="-log10(Enrichment pval)") + geom_hline(yintercept = 2)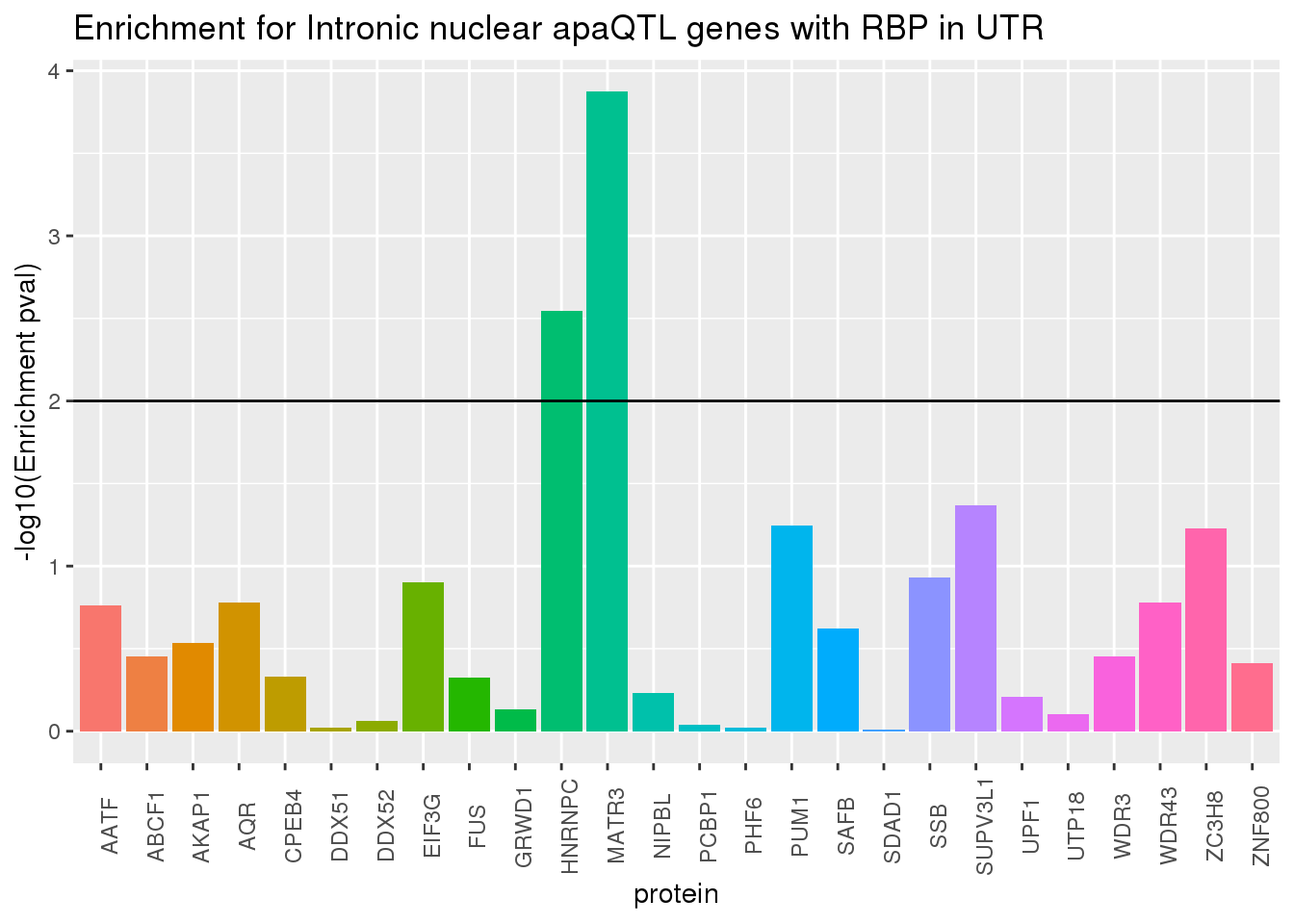
Looks like the protiens driving this are SAFB and FUS.
FUS: Associated both with RNA splicing and nuclear export.
SAFB: cotranscriptional.
Compare to total apaQTLs:
TotalQTL_genes=read.table("../data/apaQTLs/TotalapaQTLGenes.txt",col.names = "gene",stringsAsFactors = F)
TotalQTLTested_genes=read.table("../data/apaQTLs/TestedTotalapaQTLGenes.txt",col.names = "gene",stringsAsFactors = F) %>% mutate(QTL=ifelse(gene %in% QTL_genes$gene, "Yes","No"))expectedT=c()
actualT=c()
pvalT=c()
for (RBP in RBP_names$RBP){
RBPfile=read.table(paste("../data/eCLip/UTRregions_", RBP,".txt", sep=""),header=F, col.names = c('chr','start','end','gene','score','strand','RBP'), stringsAsFactors = F) %>%
inner_join(TotalQTLTested_genes, by='gene') %>%
mutate(HasRBP=ifelse(RBP!=".", "Yes","No"))
x=nrow(RBPfile %>% filter(HasRBP=="Yes", QTL=="Yes"))
m= nrow(RBPfile %>% filter(HasRBP=="Yes"))
n=nrow(RBPfile %>% filter(HasRBP!="Yes"))
k=nrow(RBPfile %>% filter(QTL=="Yes"))
expectedT=c(expectedT, which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k))))
actualT=c(actualT,x)
pvalT=c(pvalT,phyper(x,m,n,k,lower.tail=F))
}RBP_names_resT= as.data.frame(cbind(RBP=RBP_names$RBP, expectedT,actualT,pvalT)) %>% separate(RBP, into=c("exp", "protein"),sep="_")
RBP_names_resT$pvalT=as.numeric(as.character(RBP_names_resT$pvalT))
ggplot(RBP_names_resT, aes(x=protein, y=-log10(pvalT),fill=protein))+geom_bar(stat="identity") +theme(legend.position = "none", axis.text.x = element_text(angle = 90)) +labs(title="Enrichment for total apaQTL genes with RBP in UTR",y="-log10(Enrichment pval)") + geom_hline(yintercept = 2)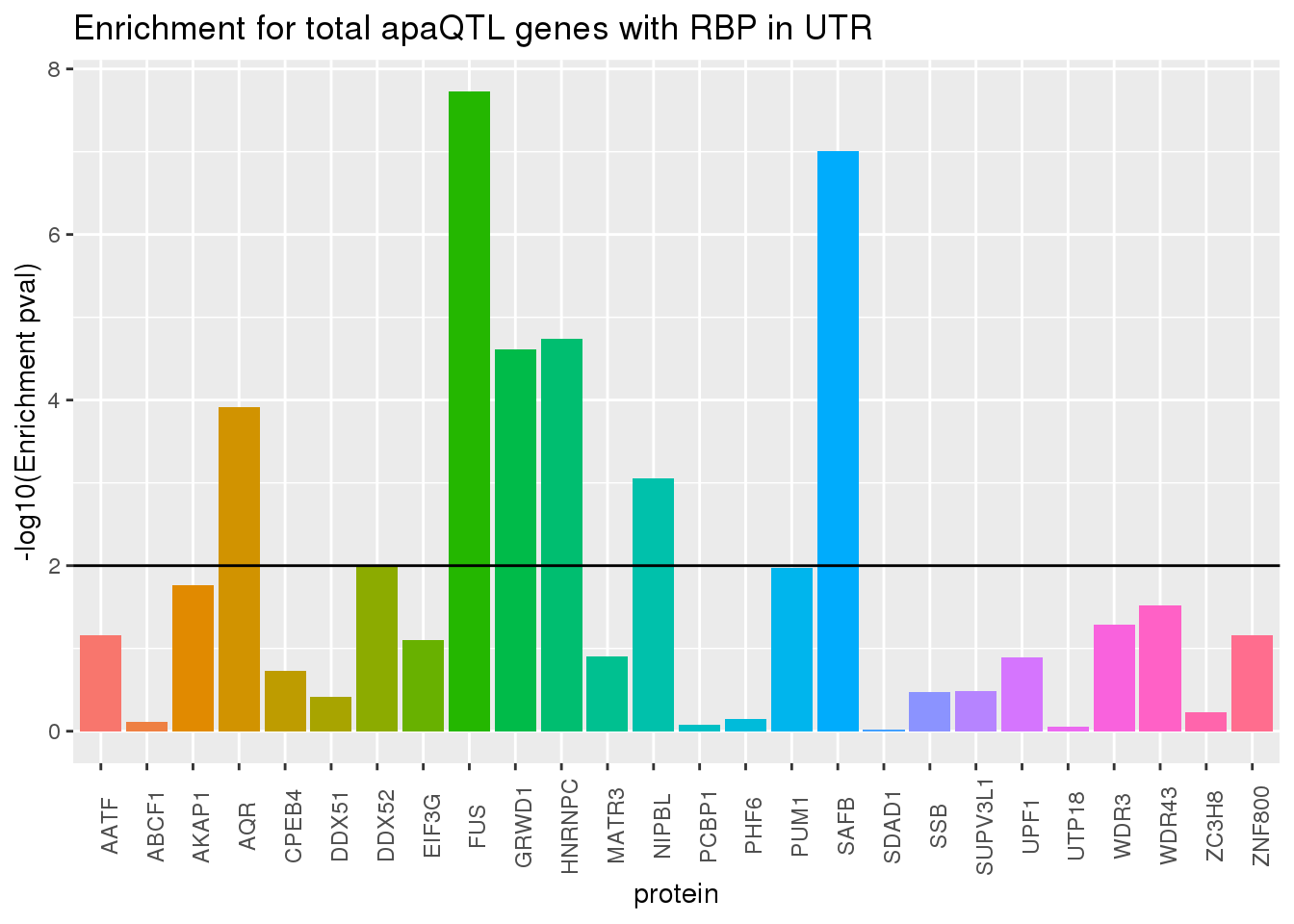
Total fraction GRWD1 and HNRNPC pop up too
GRWD1: encoded protein may play a critical role in ribosome biogenesis and may also play a role in histone methylation through interactions
HNRNPC: The hnRNPs are RNA binding proteins and they complex with heterogeneous nuclear RNA (hnRNA). These proteins are associated with pre-mRNAs in the nucleus and appear to influence pre-mRNA processing and other aspects of mRNA metabolism and transport
Linked to it:
-CPEB1 - shuttles between nucleus and the cytoplasm (Bava, F.A. et al. (2013) CPEB1 coordinates alternative 3-UTR formation with translational regulation. Nature 495, 121–125) consensus sequence 5’-UUUUUAU-3’
Will most likely have to look for binding motfis for this one.
binding disruption
Binding site disruption
Use the binding sites above
sbatch RBPdisrupt.sh NucRes=read.table("../data/eCLip/NuclearQTLoverlap_RBPbinding.txt",col.names = c("snpchr",'snpstart','snpend', 'qtl', 'dist', 'strand', 'chr','start','end', 'rbp','score', 'strndrbp'),stringsAsFactors = F)
NucRes %>% select(qtl) %>% unique() %>% nrow()[1] 37TotRes=read.table("../data/eCLip/TotalQTLoverlap_RBPbinding.txt",col.names = c("snpchr",'snpstart','snpend', 'qtl', 'dist', 'strand', 'chr','start','end', 'rbp','score', 'strndrbp'),stringsAsFactors = F)
TotRes %>% select(qtl) %>% unique() %>% nrow()[1] 2637 nuclear and 26 total qtls overlap eclip binding.
head(NucRes) snpchr snpstart snpend qtl dist strand chr
1 1 2125171 2125172 FAAP20:peak318:utr3 1830 - 1
2 1 2125171 2125172 FAAP20:peak318:utr3 1830 - 1
3 1 2125171 2125172 FAAP20:peak318:utr3 1830 - 1
4 1 6272492 6272493 RNF207:peak457:utr3 -8763 + 1
5 1 6272492 6272493 RNF207:peak457:utr3 -8763 + 1
6 1 29479003 29479004 SRSF4:peak2215:intron -14895 - 1
start end rbp score strndrbp
1 2125091 2125187 FUS_K562_rep01 200 -
2 2125111 2125192 ZNF800_K562_rep01 200 -
3 2125158 2125213 ABCF1_K562_rep01 200 -
4 6272477 6272514 AQR_K562_rep01 200 +
5 6272478 6272505 GRWD1_K562_rep01 200 +
6 29479001 29479040 UPF1_K562_rep02 200 -SRSF4 intronic QTL in a UPF1 binding site. UPF1 associated with NMD (https://www.sciencedirect.com/science/article/pii/S2211124718305837). only a qtl in the nuclear fraction.
PRR13 2 UTR variants, in UPF1 binding. total and nuclear QTL. alternative C allele associated with decreased usage of downstream isoform.
these are not significant NMD genes.
Enrichment in UTR for TvN genes
#chr10:27035787:27035907:ABI1
TvNTested=read.table("../data/DiffIso/EffectSizes.txt", header = T,stringsAsFactors = F) %>% separate(intron, into = c("chr", "start","end", "gene"),sep=":")
TvNsig=read.table("../data/highdiffsiggenes.txt",col.names = "gene", stringsAsFactors = F)
TvNTested_withinfor= TvNTested %>% select(gene) %>% unique() %>% mutate(sig=ifelse(gene %in% TvNsig$gene, "Yes", "No"))expectedTvN=c()
actualTvN=c()
pvalTvN=c()
for (RBP in RBP_names$RBP){
RBPfile=read.table(paste("../data/eCLip/UTRregions_", RBP,".txt", sep=""),header=F, col.names = c('chr','start','end','gene','score','strand','RBP'), stringsAsFactors = F) %>%
inner_join(TvNTested_withinfor, by='gene') %>%
mutate(HasRBP=ifelse(RBP!=".", "Yes","No"))
x=nrow(RBPfile %>% filter(HasRBP=="Yes", sig=="Yes"))
print(x)
m= nrow(RBPfile %>% filter(HasRBP=="Yes"))
n=nrow(RBPfile %>% filter(HasRBP!="Yes"))
k=nrow(RBPfile %>% filter(sig=="Yes"))
expectedTvN=c(expectedTvN, which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k))))
actualTvN=c(actualTvN,x)
pvalTvN=c(pvalTvN,phyper(x,m,n,k,lower.tail=F))
}[1] 115
[1] 4
[1] 417
[1] 29
[1] 269
[1] 70
[1] 222
[1] 74
[1] 230
[1] 96
[1] 143
[1] 34
[1] 456
[1] 201
[1] 331
[1] 117
[1] 307
[1] 398
[1] 28
[1] 223
[1] 37
[1] 127
[1] 102
[1] 341
[1] 122
[1] 115
[1] 4
[1] 417
[1] 29
[1] 269
[1] 70
[1] 222
[1] 74
[1] 230
[1] 96
[1] 143
[1] 34
[1] 456
[1] 201
[1] 331
[1] 117
[1] 307
[1] 398
[1] 28
[1] 223
[1] 37
[1] 127
[1] 643
[1] 102
[1] 341
[1] 122RBP_names_resTvn= as.data.frame(cbind(RBP=RBP_names$RBP, expectedTvN,actualTvN,pvalTvN)) %>% separate(RBP, into=c("exp", "protein"),sep="_")
RBP_names_resTvn$pvalTvN=as.numeric(as.character(RBP_names_resTvn$pvalTvN))
ggplot(RBP_names_resTvn, aes(x=protein, y=-log10(pvalTvN),fill=protein))+geom_bar(stat="identity") +theme(legend.position = "none", axis.text.x = element_text(angle = 90)) +labs(title="Enrichment for TvN genes with RBP in UTR",y="-log10(Enrichment pval)") + geom_hline(yintercept = 2)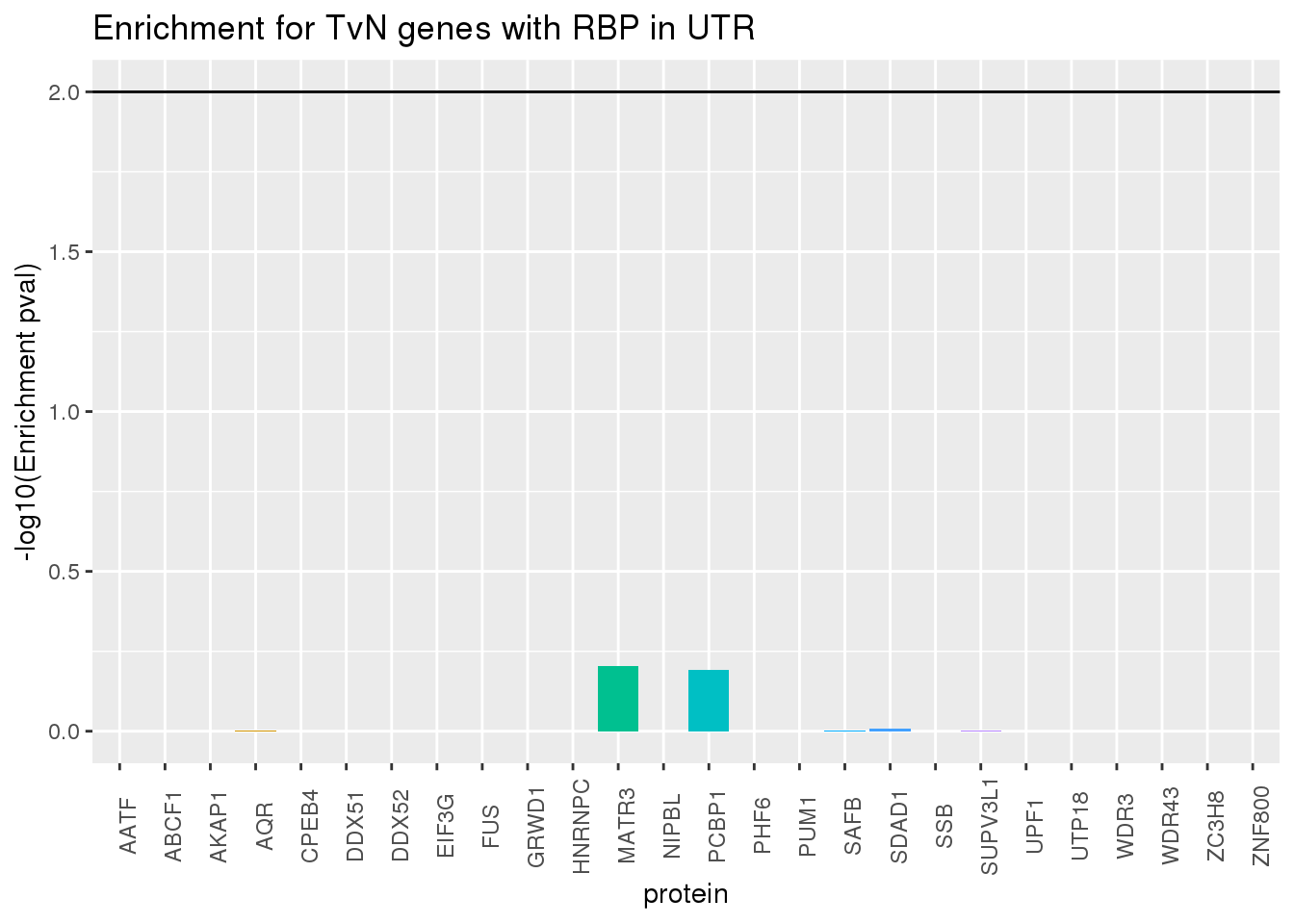
All_Tvn=read.table("../data/eCLip/AllUTRsMappedallRBP.txt",header=F, col.names = c('chr','start','end','gene','score','strand','RBP'),stringsAsFactors = F) %>%
inner_join(TvNTested_withinfor, by='gene') %>%
mutate(HasRBP=ifelse(RBP!=".", "Yes","No"))
x=nrow(All_Tvn %>% filter(HasRBP=="Yes", sig=="Yes"))
m= nrow(All_Tvn %>% filter(HasRBP=="Yes"))
n=nrow(All_Tvn %>% filter(HasRBP!="Yes"))
k=nrow(All_Tvn %>% filter(sig=="Yes"))
#expected
which(grepl(max(dhyper(1:x, m, n, k)), dhyper(1:x, m, n, k)))[1] 870#actual:
x[1] 870#pval
phyper(x,m,n,k,lower.tail=F)[1] 0.9991365RBP in UTRs does not explain TvN genes.
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] forcats_0.3.0 stringr_1.3.1 dplyr_0.8.0.1 purrr_0.3.2
[5] readr_1.3.1 tidyr_0.8.3 tibble_2.1.1 ggplot2_3.1.1
[9] tidyverse_1.2.1 workflowr_1.6.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.2 cellranger_1.1.0 plyr_1.8.4 compiler_3.5.1
[5] pillar_1.3.1 later_0.7.5 git2r_0.26.1 tools_3.5.1
[9] digest_0.6.18 lubridate_1.7.4 jsonlite_1.6 evaluate_0.12
[13] nlme_3.1-137 gtable_0.2.0 lattice_0.20-38 pkgconfig_2.0.2
[17] rlang_0.4.0 cli_1.1.0 rstudioapi_0.10 yaml_2.2.0
[21] haven_1.1.2 withr_2.1.2 xml2_1.2.0 httr_1.3.1
[25] knitr_1.20 hms_0.4.2 generics_0.0.2 fs_1.3.1
[29] rprojroot_1.3-2 grid_3.5.1 tidyselect_0.2.5 glue_1.3.0
[33] R6_2.3.0 readxl_1.1.0 rmarkdown_1.10 modelr_0.1.2
[37] magrittr_1.5 whisker_0.3-2 backports_1.1.2 scales_1.0.0
[41] promises_1.0.1 htmltools_0.3.6 rvest_0.3.2 assertthat_0.2.0
[45] colorspace_1.3-2 httpuv_1.4.5 labeling_0.3 stringi_1.2.4
[49] lazyeval_0.2.1 munsell_0.5.0 broom_0.5.1 crayon_1.3.4