Proportion eQTLs explained
Briana Mittleman
6/7/2019
Last updated: 2020-03-23
Checks: 7 0
Knit directory: apaQTL/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190411) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/.Rhistory
Ignored: data/.DS_Store
Ignored: data/ProSeq/
Ignored: output/.DS_Store
Untracked files:
Untracked: .Rprofile
Untracked: ._.DS_Store
Untracked: .gitignore
Untracked: @
Untracked: GEO_brimittleman/
Untracked: _workflowr.yml
Untracked: analysis/._PASdescriptiveplots.Rmd
Untracked: analysis/._cuttoffPercUsage.Rmd
Untracked: analysis/APApeak_Phenotype_GeneLocAnno.Nuclear.5perc.fc.gz.qqnorm.allChrom
Untracked: analysis/APApeak_Phenotype_GeneLocAnno.Total.5perc.fc.gz.qqnorm.allChrom
Untracked: analysis/QTLexampleplots.Rmd
Untracked: analysis/cuttoffPercUsage.Rmd
Untracked: analysis/eQTLoverlap.Rmd
Untracked: analysis/interpret verify bam.Rmd
Untracked: analysis/interpret_verifybam.Rmd
Untracked: analysis/mergeRNA.Rmd
Untracked: analysis/oldstuffNotNeeded.Rmd
Untracked: analysis/remove_badlines.Rmd
Untracked: analysis/totSpecInNuclear.Rmd
Untracked: analysis/totSpecIncludenotTested.Rmd
Untracked: analysis/totalspec.Rmd
Untracked: apaQTL.Rproj
Untracked: checksumsfastq.txt.gz
Untracked: code/.NascentRNAdtPlotFirstintronicPAS.sh.swp
Untracked: code/._Allsplicesite2fasta.py
Untracked: code/._ApaQTL_nominalNonnorm.sh
Untracked: code/._BothFracDTPlotGeneRegions.sh
Untracked: code/._BothFracDTPlotGeneRegions_normalized.sh
Untracked: code/._ClosestTissuePAS.sh
Untracked: code/._ColocApAeQTL.sh
Untracked: code/._ColocApAeQTL_PM.sh
Untracked: code/._Coloc_generalAPAeQTL.R
Untracked: code/._Coloc_generalAPAeQTL_PM.R
Untracked: code/._CreateRNALZforeQTLs.sh
Untracked: code/._CreateRNALZnucAPAqtls.sh
Untracked: code/._DistPAS2Sig_RandomIntron.py
Untracked: code/._EandPqtl_perm.sh
Untracked: code/._EandPqtls.sh
Untracked: code/._ExtractGene4eQTLLZ.py
Untracked: code/._ExtractGene4eQTLLZpy
Untracked: code/._ExtractGeneRNAAssoc.py
Untracked: code/._ExtractPAS4LZeQTLs.py
Untracked: code/._ExtractPAS4eQTLsLZ.sh
Untracked: code/._ExtractPASforLZ.py
Untracked: code/._ExtractPASforLZ_run.sh
Untracked: code/._FC_NucintornUpandDown.sh
Untracked: code/._FC_UTR.sh
Untracked: code/._FC_intornUpandDownsteamPAS.sh
Untracked: code/._FC_nascentseq.sh
Untracked: code/._FC_newPeaks_olddata.sh
Untracked: code/._HMMpermuteTotal.py
Untracked: code/._HmmPermute.py
Untracked: code/._IntronicPASDT.sh
Untracked: code/._LC_samplegroups.py
Untracked: code/._LD_qtl.sh
Untracked: code/._LD_snpsproxy.sh
Untracked: code/._MapAllRBP.sh
Untracked: code/._NascentRNAdtPlot.sh
Untracked: code/._NascentRNAdtPlot3UTRPAS.sh
Untracked: code/._NascentRNAdtPlotExcludeFirstintronicPAS.sh
Untracked: code/._NascentRNAdtPlotNucPAS.sh
Untracked: code/._NascentRNAdtPlotTotPAS.sh
Untracked: code/._NascentRNAdtPlotintronicPAS.sh
Untracked: code/._NascnetRNAdtPlotPAS.sh
Untracked: code/._NetSeq_fourthintronDT.sh
Untracked: code/._NomResfromPASSNP.py
Untracked: code/._NuclearPAS_5per.bed.py
Untracked: code/._NuclearandRNA5samp_dtplots.sh
Untracked: code/._PTTfacetboxplots.R
Untracked: code/._PrematureQTLNominal.sh
Untracked: code/._PrematureQTLPermuted.sh
Untracked: code/._QTL2bed.py
Untracked: code/._QTL2bed_withstrand.py
Untracked: code/._RBPdisrupt.sh
Untracked: code/._RNAbam2bw.sh
Untracked: code/._RNAseqDTplot.sh
Untracked: code/._Randomsplicesite2fasta.py
Untracked: code/._Rplots.pdf
Untracked: code/._RunRes2PAS.sh
Untracked: code/._SAF215upbed.py
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilefiltPAS
Untracked: code/._TESplots100bp.sh
Untracked: code/._TESplots150bp.sh
Untracked: code/._TESplots200bp.sh
Untracked: code/._TotalPAS_5perc.bed.py
Untracked: code/._Totspec_example.sh
Untracked: code/._Totspec_exampleTOT.sh
Untracked: code/._Untitled
Untracked: code/._ZipandTabPheno.sh
Untracked: code/._aAPAqtl_nominal39ind.sh
Untracked: code/._allNucSpecQTLine.py
Untracked: code/._allNucSpecfromNonNorm.py
Untracked: code/._annotatePacBioPASregion.sh
Untracked: code/._annotatedPAS2bed.py
Untracked: code/._apaInPandE.py
Untracked: code/._apaQTLCorrectPvalMakeQQ.R
Untracked: code/._apaQTLCorrectpval_6or7a.R
Untracked: code/._apaQTL_Nominal.sh
Untracked: code/._apaQTL_nominalInclusive.sh
Untracked: code/._apaQTL_nominalv67.sh
Untracked: code/._apaQTL_permuted.sh
Untracked: code/._apaQTL_permuted_test6A7A.sh
Untracked: code/._apainRibo.py
Untracked: code/._assignNucIntonpeak2intronlocs.sh
Untracked: code/._assignTotIntronpeak2intronlocs.sh
Untracked: code/._bam2BW_5primemost.sh
Untracked: code/._bed2saf.py
Untracked: code/._bothFracDTplot1stintron.sh
Untracked: code/._bothFracDTplot4thintron.sh
Untracked: code/._bothFrac_FC.sh
Untracked: code/._callPeaksYL.py
Untracked: code/._changeRibonomQTLres2genename.py
Untracked: code/._changenomQTLres2geneName.py
Untracked: code/._chooseAnno2PAS_pacbio.py
Untracked: code/._chooseAnno2SAF.py
Untracked: code/._chooseSignalSite
Untracked: code/._chooseSignalSite.py
Untracked: code/._closestannotated.sh
Untracked: code/._closestannotated_byfrac.sh
Untracked: code/._cluster.json
Untracked: code/._clusterPAS.json
Untracked: code/._clusterfiltPAS.json
Untracked: code/._codingdms2bed.py
Untracked: code/._config.yaml
Untracked: code/._config2.yaml
Untracked: code/._configOLD.yaml
Untracked: code/._convertNominal2SNPLOC.py
Untracked: code/._convertNominal2SNPloc2Versions.py
Untracked: code/._convertNumeric.py
Untracked: code/._correctNomeqtl.R
Untracked: code/._createPlinkSampfile.py
Untracked: code/._dag.pdf
Untracked: code/._eQTL_switch2snploc.py
Untracked: code/._eQTLgenestestedapa.py
Untracked: code/._encodeRNADTplots.sh
Untracked: code/._extactPAS100meanphyloP.py
Untracked: code/._extractGeneLZfiles.sh
Untracked: code/._extractGeneLZfileseQTLs.sh
Untracked: code/._extractGenotypes.py
Untracked: code/._extractPACmeanPhyloP.py
Untracked: code/._extractPhylop50up.py
Untracked: code/._extractPhylopextra50.py
Untracked: code/._extractRNApval4lz.py
Untracked: code/._extractseqfromqtlfastq.py
Untracked: code/._fc2leafphen.py
Untracked: code/._fc_filteredPAS6and7As.sh
Untracked: code/._fifteenBPupstreamPAS.py
Untracked: code/._fiftyBPupstreamPAS.py
Untracked: code/._filter5perc.R
Untracked: code/._filter5percPheno.py
Untracked: code/._filterLDsnps.py
Untracked: code/._filterMPPAS.py
Untracked: code/._filterMPPAS_15.py
Untracked: code/._filterMPPAS_15_7As.py
Untracked: code/._filterMPPAS_50.py
Untracked: code/._filterSAFforMP.py
Untracked: code/._filterpeaks.py
Untracked: code/._finalPASbed2SAF.py
Untracked: code/._fix4su304corr.py
Untracked: code/._fix4su604corr.py
Untracked: code/._fix4sukalisto.py
Untracked: code/._fixExandUnexeQTL
Untracked: code/._fixExandUnexeQTL.py
Untracked: code/._fixFChead.py
Untracked: code/._fixFChead_bothfrac.py
Untracked: code/._fixFChead_short.py
Untracked: code/._fixGWAS4Munge.py
Untracked: code/._fixH3k12ac.py
Untracked: code/._fixPASregionSNPs.py
Untracked: code/._fixRNAhead4corr.py
Untracked: code/._fixRNAkalisto.py
Untracked: code/._fix_randomIntron.py
Untracked: code/._fixgroupedtranscript.py
Untracked: code/._fixhead_netseqfc.py
Untracked: code/._getAPAfromanyeQTL.py
Untracked: code/._getApapval4eqtl.py
Untracked: code/._getApapval4eqtl_unexp.py
Untracked: code/._getApapval4eqtl_version67.py
Untracked: code/._getDownstreamIntronNuclear.py
Untracked: code/._getIntronDownstreamPAS.py
Untracked: code/._getIntronUpstreamPAS.py
Untracked: code/._getQTLalleles.py
Untracked: code/._getQTLfastq.sh
Untracked: code/._getUpstreamIntronNuclear.py
Untracked: code/._grouptranscripts.py
Untracked: code/._intersectVCFandupPAS.sh
Untracked: code/._keep5perMAF.py
Untracked: code/._keepSNP_vcf.sh
Untracked: code/._make5percPeakbed.py
Untracked: code/._makeFileID.py
Untracked: code/._makePheno.py
Untracked: code/._makeSAFbothfrac5perc.py
Untracked: code/._makeSNP2rsidfile.py
Untracked: code/._makeeQTLempirical_unexp.py
Untracked: code/._makeeQTLempiricaldist.py
Untracked: code/._makegencondeTSSfile.py
Untracked: code/._mapSSsnps2PAS.sh
Untracked: code/._mergRNABam.sh
Untracked: code/._mergeAllBam.sh
Untracked: code/._mergeAnnotations.sh
Untracked: code/._mergeBW_norm.sh
Untracked: code/._mergeBamNascent.sh
Untracked: code/._mergeByFracBam.sh
Untracked: code/._mergePeaks.sh
Untracked: code/._miRNAdisrupt.sh
Untracked: code/._mnase1stintron.sh
Untracked: code/._mnaseDT_fourthintron.sh
Untracked: code/._namePeaks.py
Untracked: code/._netseqDTplot1stIntron.sh
Untracked: code/._netseqFC.sh
Untracked: code/._nominavalfortotspec.py
Untracked: code/._noninalpval4alltot.py
Untracked: code/._nucQTLGWAS.py
Untracked: code/._nucSpecQTLineData.py
Untracked: code/._nucSpeceffectsize.py
Untracked: code/._nucspecnucPASine.py
Untracked: code/._pQTLsotherdata.py
Untracked: code/._pacbioDT.sh
Untracked: code/._pacbioIntronicDT.sh
Untracked: code/._parseALLSSres.py
Untracked: code/._parseBestbamid.py
Untracked: code/._parseLDRes.py
Untracked: code/._parseLDresBothPAS.sh
Untracked: code/._parseRanodmSSres.py
Untracked: code/._parseSSres.py
Untracked: code/._peak2PAS.py
Untracked: code/._peakFC.sh
Untracked: code/._pheno2countonly.R
Untracked: code/._phenoQTLfromlist.py
Untracked: code/._processYRIgen.py
Untracked: code/._pttQTLsinapaQTL.py
Untracked: code/._qtlRegionseq.sh
Untracked: code/._qtlsPvalOppFrac.py
Untracked: code/._quantassign2parsedpeak.py
Untracked: code/._removeXfromHmm.py
Untracked: code/._removeloc_pheno.py
Untracked: code/._riboQTL.sh
Untracked: code/._runCorrectNomEqtl.sh
Untracked: code/._runFixGWAS4Munge.sh
Untracked: code/._runHMMpermuteAPAqtls.sh
Untracked: code/._runHMMpermuteeQTLS.sh
Untracked: code/._runMakeEmpiricaleQTL_unexp.sh
Untracked: code/._runMakeeQTLempirical.sh
Untracked: code/._run_bam2bw_all3prime.sh
Untracked: code/._run_bam2bw_extra3.sh
Untracked: code/._run_bestbamid.sj
Untracked: code/._run_dist2sig_randomintron.sh
Untracked: code/._run_filtersnpLD.sh
Untracked: code/._run_getAPAfromeQTL_version6.7.sh
Untracked: code/._run_getApaPval4eqtl.sh
Untracked: code/._run_getapafromeQTL.py
Untracked: code/._run_getapafromeQTL.sh
Untracked: code/._run_getapapval4eqtl_unexp.sh
Untracked: code/._run_leafcutterDiffIso.sh
Untracked: code/._run_prxySNP.sh
Untracked: code/._run_pttfacetboxplot.sh
Untracked: code/._run_sepUsagephen.sh
Untracked: code/._run_sepgenobychrom.sh
Untracked: code/._run_verifybam.sh
Untracked: code/._selectNominalPvalues.py
Untracked: code/._sepUsagePhen.py
Untracked: code/._sepgenobychrom.py
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._sortindexRNAbam.sh
Untracked: code/._specAPAinE.py
Untracked: code/._splicesite2fasta.py
Untracked: code/._submit-snakemakePAS.sh
Untracked: code/._submit-snakemakefiltPAS.sh
Untracked: code/._subsetAPAnotEorPgene.py
Untracked: code/._subsetAPAnotEorPgene_2versions.py
Untracked: code/._subsetAPAnotEorR.py
Untracked: code/._subsetApanoteGene.py
Untracked: code/._subsetApanoteGene_2versions.py
Untracked: code/._subsetNootherQTL.py
Untracked: code/._subsetUnexplainedeQTLs.py
Untracked: code/._subsetVCF_SS.sh
Untracked: code/._subsetVCF_noSSregions.sh
Untracked: code/._subsetVCF_upstreamPAS.sh
Untracked: code/._subset_diffisopheno.py
Untracked: code/._subsetpermAPAwithGenelist.py
Untracked: code/._subsetpermAPAwithGenelist_2versions.py
Untracked: code/._subsetvcf_otherreg.sh
Untracked: code/._subsetvcf_permSS.sh
Untracked: code/._subtrachfiveprimeUTR.sh
Untracked: code/._subtractExons.sh
Untracked: code/._subtractfiveprimeUTR.sh
Untracked: code/._tabixSNPS.sh
Untracked: code/._tenBPupstreamPAS.py
Untracked: code/._test.pdf
Untracked: code/._testVerifyBam.sh
Untracked: code/._tissuePAS2hg19.sh
Untracked: code/._totSeceffectsize.py
Untracked: code/._totspecinE.py
Untracked: code/._totspecqtlFacetBoxplots.sh
Untracked: code/._totspecqtlFacetBoxplotsTOT.sh
Untracked: code/._twentyBPupstreamPAS.py
Untracked: code/._utrdms2saf.py
Untracked: code/._vcf2bed.py
Untracked: code/._verifyBam18517N.sh
Untracked: code/._verifyBam18517T.sh
Untracked: code/._verifyBam19128N.sh
Untracked: code/._verifyBam19128T.sh
Untracked: code/._wrap_verifybam.sh
Untracked: code/._writePTTexamplecode.py
Untracked: code/._writePTTexamplecode.sh
Untracked: code/.pversion
Untracked: code/.snakemake/
Untracked: code/1
Untracked: code/APAqtl_nominal.err
Untracked: code/APAqtl_nominal.out
Untracked: code/APAqtl_nominal_39.err
Untracked: code/APAqtl_nominal_39.out
Untracked: code/APAqtl_nominal_inclusive.err
Untracked: code/APAqtl_nominal_inclusive.out
Untracked: code/APAqtl_nominal_nonNorm.err
Untracked: code/APAqtl_nominal_nonNorm.out
Untracked: code/APAqtl_nominal_versions67.err
Untracked: code/APAqtl_nominal_versions67.out
Untracked: code/APAqtl_permuted.err
Untracked: code/APAqtl_permuted.out
Untracked: code/APAqtl_permuted_versions67.err
Untracked: code/APAqtl_permuted_versions67.out
Untracked: code/Allsplicesite2fasta.py
Untracked: code/BothFracDTPlot1stintron.err
Untracked: code/BothFracDTPlot1stintron.out
Untracked: code/BothFracDTPlot4stintron.err
Untracked: code/BothFracDTPlot4stintron.out
Untracked: code/BothFracDTPlotGeneRegions.err
Untracked: code/BothFracDTPlotGeneRegions.out
Untracked: code/BothFracDTPlotGeneRegions_norm.err
Untracked: code/BothFracDTPlotGeneRegions_norm.out
Untracked: code/ClosestTissuePAS.sh
Untracked: code/ColocApAeQTL.err
Untracked: code/ColocApAeQTL.out
Untracked: code/ColocApAeQTL.sh
Untracked: code/ColocApAeQTLPM.err
Untracked: code/ColocApAeQTLPM.out
Untracked: code/ColocApAeQTL_PM.sh
Untracked: code/Coloc_generalAPAeQTL.R
Untracked: code/Coloc_generalAPAeQTL_PM.R
Untracked: code/CreateRNALZforeQTLs.sh
Untracked: code/CreateRNALZnucAPAqtls.sh
Untracked: code/DistPAS2Sig_RandomIntron.py
Untracked: code/EandPqtl.err
Untracked: code/EandPqtl.out
Untracked: code/EncodeRNADTPlotGeneRegions.err
Untracked: code/EncodeRNADTPlotGeneRegions.out
Untracked: code/ExtractGene4eQTLLZ.py
Untracked: code/ExtractGene4eQTLLZpy
Untracked: code/ExtractGeneRNAAssoc.py
Untracked: code/ExtractPAS4LZeQTLs.py
Untracked: code/ExtractPAS4eQTLsLZ.sh
Untracked: code/ExtractPASforLZ.py
Untracked: code/ExtractPASforLZ_run.sh
Untracked: code/FC_NucintronPASupandDown.err
Untracked: code/FC_NucintronPASupandDown.out
Untracked: code/FC_UTR.err
Untracked: code/FC_UTR.out
Untracked: code/FC_intronPASupandDown.err
Untracked: code/FC_intronPASupandDown.out
Untracked: code/FC_nascent.err
Untracked: code/FC_nascentout
Untracked: code/FC_newPAS_olddata.err
Untracked: code/FC_newPAS_olddata.out
Untracked: code/HmmPermute.p
Untracked: code/IntronicPASDT.err
Untracked: code/IntronicPASDT.out
Untracked: code/LD_vcftools.hap.out
Untracked: code/MapAllRBP.sh
Untracked: code/MapRBP.err
Untracked: code/MapRBP.out
Untracked: code/NascentDTPlotGeneRegions.err
Untracked: code/NascentDTPlotGeneRegions.out
Untracked: code/NascentDTPlotPAS.err
Untracked: code/NascentDTPlotPAS.out
Untracked: code/NascentDTPlotPAS_3utr.err
Untracked: code/NascentDTPlotPAS_3utr.out
Untracked: code/NascentDTPlotPAS_firstintron.err
Untracked: code/NascentDTPlotPAS_firstintron.out
Untracked: code/NascentDTPlotPAS_intron.err
Untracked: code/NascentDTPlotPAS_intron.out
Untracked: code/NascentDTPlotPAS_nuc.err
Untracked: code/NascentDTPlotPAS_nuc.out
Untracked: code/NascentDTPlotPAS_tot.err
Untracked: code/NascentDTPlotPAS_tot.out
Untracked: code/Nuclear_example.err
Untracked: code/Nuclear_example.out
Untracked: code/NuclearandRNA5samp_dtplots.sh
Untracked: code/NuclearandRNAFracDTPlotGeneRegions.err
Untracked: code/NuclearandRNAFracDTPlotGeneRegions.out
Untracked: code/PACbioDT.err
Untracked: code/PACbioDT.out
Untracked: code/PACbioDTitronic.err
Untracked: code/PACbioDTitronic.out
Untracked: code/Prematureqtl_nominal.err
Untracked: code/Prematureqtl_nominal.out
Untracked: code/Prematureqtl_permuted.err
Untracked: code/Prematureqtl_permuted.out
Untracked: code/RBPdisrupt.err
Untracked: code/RBPdisrupt.out
Untracked: code/RBPdisrupt.sh
Untracked: code/README.md
Untracked: code/RNABam2BW.err
Untracked: code/RNABam2BW.out
Untracked: code/RNAseqDTPlotGeneRegions.err
Untracked: code/RNAseqDTPlotGeneRegions.out
Untracked: code/Randomsplicesite2fasta.py
Untracked: code/Rplots.pdf
Untracked: code/TESplots100bp.err
Untracked: code/TESplots100bp.out
Untracked: code/TESplots150bp.err
Untracked: code/TESplots150bp.out
Untracked: code/TESplots200bp.err
Untracked: code/TESplots200bp.out
Untracked: code/Tissueclosestannotated.err
Untracked: code/Tissueclosestannotated.out
Untracked: code/Total_example.err
Untracked: code/Total_example.out
Untracked: code/Totspec_example.err
Untracked: code/Totspec_example.out
Untracked: code/Totspec_example.sh
Untracked: code/Totspec_exampleTOT.err
Untracked: code/Totspec_exampleTOT.out
Untracked: code/Totspec_exampleTOT.sh
Untracked: code/Untitled
Untracked: code/YRI_LCL.vcf.gz
Untracked: code/YRI_LCL_chr1.vcf.gz.log
Untracked: code/YRI_LCL_chr1.vcf.gz.recode.vcf
Untracked: code/annotatedPASregion.err
Untracked: code/annotatedPASregion.out
Untracked: code/apaQTL_nominalInclusive.sh
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assigntotPeak2Intronicregion.err
Untracked: code/assigntotPeak2Intronicregion.out
Untracked: code/bam2bw.err
Untracked: code/bam2bw.out
Untracked: code/bam2bw_5primemost.err
Untracked: code/bam2bw_5primemost.out
Untracked: code/binary_fileset.log
Untracked: code/bothFrac_FC.err
Untracked: code/bothFrac_FC.out
Untracked: code/callSHscripts.txt
Untracked: code/closestannotated.err
Untracked: code/closestannotated.out
Untracked: code/closestannotatedbyfrac.err
Untracked: code/closestannotatedbyfrac.out
Untracked: code/dag.pdf
Untracked: code/dagPAS.pdf
Untracked: code/dagfiltPAS.pdf
Untracked: code/extactPAS100meanphyloP.py
Untracked: code/extractGeneLZfiles.err
Untracked: code/extractGeneLZfiles.out
Untracked: code/extractGeneLZfiles.sh
Untracked: code/extractGeneLZfileseQTLs.err
Untracked: code/extractGeneLZfileseQTLs.out
Untracked: code/extractGeneLZfileseQTLs.sh
Untracked: code/extractPACmeanPhyloP.py
Untracked: code/extractPASLZfiles.err
Untracked: code/extractPASLZfiles.out
Untracked: code/extractPASLZfileseQTLs.err
Untracked: code/extractPASLZfileseQTLs.out
Untracked: code/extractPhylop50up.py
Untracked: code/extractPhylopextra50.py
Untracked: code/extractRNApval4lz.py
Untracked: code/fixExandUnexeQTL
Untracked: code/fixGWAS4Munge.py
Untracked: code/fix_randomIntron.py
Untracked: code/fixmunge
Untracked: code/genotypesYRI.gen.proc.keep.vcf.log
Untracked: code/genotypesYRI.gen.proc.keep.vcf.recode.vcf
Untracked: code/getseq100up.err
Untracked: code/getseq100up.out
Untracked: code/grouptranscripts.err
Untracked: code/grouptranscripts.out
Untracked: code/intersectPAS_ssSNPS.err
Untracked: code/intersectPAS_ssSNPS.out
Untracked: code/intersectVCFPAS.err
Untracked: code/intersectVCFPAS.out
Untracked: code/liftoverPAShg38to19.err
Untracked: code/liftoverPAShg38to19.out
Untracked: code/log/
Untracked: code/logs/
Untracked: code/merge53PRIMEbam.err
Untracked: code/merge53PRIMEbam.out
Untracked: code/merge53RNAbam.err
Untracked: code/merge53prime.sh
Untracked: code/merge5RNABam.err
Untracked: code/merge5RNABam.out
Untracked: code/merge5RNAbam.out
Untracked: code/merge5RNAbam.sh
Untracked: code/mergeAnno.err
Untracked: code/mergeAnno.out
Untracked: code/mergeBWnorm.err
Untracked: code/mergeBWnorm.out
Untracked: code/mergeBamNacent.err
Untracked: code/mergeBamNacent.out
Untracked: code/mergeRNAbam.err
Untracked: code/mergeRNAbam.out
Untracked: code/miRNAdisrupt.err
Untracked: code/miRNAdisrupt.out
Untracked: code/miRNAdisrupt.sh
Untracked: code/mnaseDTPlot1stintron.err
Untracked: code/mnaseDTPlot1stintron.out
Untracked: code/mnaseDTPlot4thintron.err
Untracked: code/mnaseDTPlot4thintron.out
Untracked: code/netDTPlot4thintron.out
Untracked: code/netseqFC.err
Untracked: code/netseqFC.out
Untracked: code/neyDTPlot4thintron.err
Untracked: code/nominavalfortotspec.py
Untracked: code/noninalpval4alltot.py
Untracked: code/nucspecinE.py
Untracked: code/parseALLSSres.py
Untracked: code/parseLDRes.py
Untracked: code/parseLDres.err
Untracked: code/parseLDres.out
Untracked: code/parseLDresBothPAS.sh
Untracked: code/parseRanodmSSres.py
Untracked: code/parseSSres.py
Untracked: code/plink.log
Untracked: code/prxySNP.err
Untracked: code/prxySNP.out
Untracked: code/pttFacetBoxplots.err
Untracked: code/pttFacetBoxplots.out
Untracked: code/qtlFacetBoxplots.err
Untracked: code/qtlFacetBoxplots.out
Untracked: code/rLD_vcftools.hap.err
Untracked: code/riboqtl.err
Untracked: code/riboqtl.out
Untracked: code/runBestBamID.err
Untracked: code/runCorrectNomeqtl.err
Untracked: code/runCorrectNomeqtl.out
Untracked: code/runFilterLD.err
Untracked: code/runFilterLD.out
Untracked: code/runFixGWAS4Munge.sh
Untracked: code/runHMMpermute.err
Untracked: code/runHMMpermute.out
Untracked: code/runHMMpermuteeQTLs.err
Untracked: code/runHMMpermuteeQTLs.out
Untracked: code/runMakeEmpiricaleQTLs.err
Untracked: code/runMakeEmpiricaleQTLs.out
Untracked: code/runMakeEmpiricaleQTLsunex.err
Untracked: code/runMakeEmpiricaleQTLsunex.out
Untracked: code/run_DistPAS2Sig.err
Untracked: code/run_DistPAS2Sig.out
Untracked: code/run_DistPAS2Sig_intron.err
Untracked: code/run_DistPAS2Sig_intron.out
Untracked: code/run_bam2bw.err
Untracked: code/run_bam2bw.out
Untracked: code/run_bam2bwexta.err
Untracked: code/run_bam2bwexta.out
Untracked: code/run_dist2sig_randomintron.sh
Untracked: code/run_getAPAfromanyeQTL.err
Untracked: code/run_getAPAfromanyeQTL.out
Untracked: code/run_getApaPval4eQTLs.err
Untracked: code/run_getApaPval4eQTLs.out
Untracked: code/run_getApaPval4eQTLsunexplained.err
Untracked: code/run_getApaPval4eQTLsunexplained.out
Untracked: code/run_leafcutter_ds.err
Untracked: code/run_leafcutter_ds.out
Untracked: code/run_sepgenobychrom.err
Untracked: code/run_sepgenobychrom.out
Untracked: code/run_sepusage.err
Untracked: code/run_sepusage.out
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/run_verifybam128N.err
Untracked: code/run_verifybam128N.out
Untracked: code/run_verifybam128T.err
Untracked: code/run_verifybam128T.out
Untracked: code/run_verifybam517N.err
Untracked: code/run_verifybam517N.out
Untracked: code/run_verifybam517T.err
Untracked: code/run_verifybam517T.out
Untracked: code/runprxySNP.err
Untracked: code/runprxySNP.out
Untracked: code/runres2pas.err
Untracked: code/runres2pas.out
Untracked: code/scripts/
Untracked: code/scripts_PAS_500_Lymph/
Untracked: code/seqQTLfastq.err
Untracked: code/seqQTLfastq.out
Untracked: code/seqQTLregion.err
Untracked: code/seqQTLregion.out
Untracked: code/snakePASlog.out
Untracked: code/snakefiltPASlog.out
Untracked: code/sortindexRNABam.err
Untracked: code/sortindexRNABam.out
Untracked: code/specAPAinE.py
Untracked: code/splicesite2fasta.py
Untracked: code/subsetAPAnotEorR.py
Untracked: code/subsetNootherQTL.py
Untracked: code/subsetvcf_SS.err
Untracked: code/subsetvcf_SS.out
Untracked: code/subsetvcf_noSS.err
Untracked: code/subsetvcf_noSS.out
Untracked: code/subsetvcf_pas.err
Untracked: code/subsetvcf_pas.out
Untracked: code/subsetvcf_perm.err
Untracked: code/subsetvcf_perm.out
Untracked: code/subsetvcf_rand.err
Untracked: code/subsetvcf_rand.out
Untracked: code/subtract5UTR.err
Untracked: code/subtract5UTR.out
Untracked: code/subtractExons.err
Untracked: code/subtractExons.out
Untracked: code/tabixSNPs.err
Untracked: code/tabixSNPs.out
Untracked: code/test.pdf
Untracked: code/testFix.txt
Untracked: code/test_verifybam.err
Untracked: code/test_verifybam.out
Untracked: code/tissuePAS2hg19.sh
Untracked: code/totspecinE.py
Untracked: code/totspecqtlFacetBoxplots.err
Untracked: code/totspecqtlFacetBoxplots.out
Untracked: code/totspecqtlFacetBoxplots.sh
Untracked: code/totspecqtlFacetBoxplotsTOT.err
Untracked: code/totspecqtlFacetBoxplotsTOT.out
Untracked: code/totspecqtlFacetBoxplotsTOT.sh
Untracked: code/vcf_keepsnps.err
Untracked: code/vcf_keepsnps.out
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/zipandtabPhen.err
Untracked: code/zipandtabPhen.out
Untracked: data/._.DS_Store
Untracked: data/._MetaDataSequencing.txt
Untracked: data/AnnotatedPAS/
Untracked: data/ApaByEgene/
Untracked: data/ApaByPgene/
Untracked: data/ApaByRgene/
Untracked: data/BadLines/
Untracked: data/BaseComp/
Untracked: data/Battle_pQTL/
Untracked: data/CheckSums/
Untracked: data/CompareOldandNew/
Untracked: data/DTmatrix/
Untracked: data/DiffIso/
Untracked: data/EncodeRNA/
Untracked: data/ExampleQTLPlots/
Untracked: data/ExampleQTLPlots_update/
Untracked: data/ExpressionIndependentapaQTLs.txt
Untracked: data/FiveMergedBW/
Untracked: data/FiveMergedBam/
Untracked: data/FlaggedPAS/
Untracked: data/GWAS_overlap/
Untracked: data/Geuvadis/
Untracked: data/GeuvadisRNA/
Untracked: data/GeuvadiseQTL/
Untracked: data/HMMqtls/
Untracked: data/LDSR_annotations/
Untracked: data/LZ_both/
Untracked: data/Li_eQTLs/
Untracked: data/NMD/
Untracked: data/NascentRNA/
Untracked: data/NucSpeceQTLeffect/
Untracked: data/PAS/
Untracked: data/PAS_postFlag/
Untracked: data/PolyA_DB/
Untracked: data/PreTerm_pheno/
Untracked: data/PrematureQTLNominal/
Untracked: data/PrematureQTLPermuted/
Untracked: data/QTLGenotypes/
Untracked: data/QTLoverlap/
Untracked: data/QTLoverlap_inclusive/
Untracked: data/QTLoverlap_nonNorm/
Untracked: data/README.md
Untracked: data/RNAseq/
Untracked: data/Reads2UTR/
Untracked: data/SNPinSS/
Untracked: data/SignalSiteFiles/
Untracked: data/TF_motifdisruption/
Untracked: data/TSS/
Untracked: data/ThirtyNineIndQtl_nominal/
Untracked: data/TissueData/
Untracked: data/Version15bp6As/
Untracked: data/Version15bp7As/
Untracked: data/apaQTLNominal/
Untracked: data/apaQTLNominal_4pc/
Untracked: data/apaQTLNominal_inclusive/
Untracked: data/apaQTLPermuted/
Untracked: data/apaQTLPermuted_4pc/
Untracked: data/apaQTLs/
Untracked: data/assignedPeaks/
Untracked: data/assignedPeaks_15Up/
Untracked: data/bam/
Untracked: data/bam_clean/
Untracked: data/bam_waspfilt/
Untracked: data/bed_10up/
Untracked: data/bed_clean/
Untracked: data/bed_clean_sort/
Untracked: data/bed_waspfilter/
Untracked: data/bedsort_waspfilter/
Untracked: data/bothFrac_FC/
Untracked: data/bw/
Untracked: data/bw_norm/
Untracked: data/coloc/
Untracked: data/coloc_PM/
Untracked: data/eCLip/
Untracked: data/eQTL_LZ/
Untracked: data/eQTLs/
Untracked: data/exampleQTLs/
Untracked: data/exosome/
Untracked: data/fastq/
Untracked: data/filterPeaks/
Untracked: data/fourSU/
Untracked: data/h3k27ac/
Untracked: data/highdiffsiggenes.txt
Untracked: data/inclusivePeaks/
Untracked: data/inclusivePeaks_FC/
Untracked: data/intronRNAratio/
Untracked: data/intron_analysis/
Untracked: data/locusZoom/
Untracked: data/mergedBG/
Untracked: data/mergedBW_byfrac/
Untracked: data/mergedBW_norm/
Untracked: data/mergedBam/
Untracked: data/mergedbyFracBam/
Untracked: data/miRNAbinding/
Untracked: data/molPhenos/
Untracked: data/molQTLs/
Untracked: data/motifdistrupt/
Untracked: data/nPAS/
Untracked: data/netseq/
Untracked: data/nonNorm_pheno/
Untracked: data/nuc_10up/
Untracked: data/nuc_10upclean/
Untracked: data/oldPASfiles/
Untracked: data/overlapeQTL_try2/
Untracked: data/overlapeQTLs/
Untracked: data/pQTLoverlap/
Untracked: data/pacbio/
Untracked: data/peakCoverage/
Untracked: data/peaks_5perc/
Untracked: data/phenotype/
Untracked: data/phenotype_5perc/
Untracked: data/phenotype_inclusivePAS/
Untracked: data/phylop/
Untracked: data/pttQTL/
Untracked: data/pttQTLplots/
Untracked: data/sigDiffGenes.txt
Untracked: data/sort/
Untracked: data/sort_clean/
Untracked: data/sort_waspfilter/
Untracked: data/splicesite/
Untracked: data/totSpecExampleQTLPlots/
Untracked: data/totSpecExampleQTLPlots_tot/
Untracked: data/twoMech/
Untracked: data/vareQTLvarAPAqtl/
Untracked: data/verifyBAM/
Untracked: data/verifyBAM_full/
Untracked: nohup.out
Untracked: output/._.DS_Store
Untracked: output/._AverageDiffHeatmap.Nuclear.png
Untracked: output/._AverageDiffHeatmap.Total.png
Untracked: output/._GeneswithAPApotential.png
Untracked: output/._GeneswithAPApotentialAllPAS.png
Untracked: output/._PASlocation.png
Untracked: output/._SignalSitePlot.png
Untracked: output/._meanCorrelationPhenotypes.svg
Untracked: output/._qqplot_Nuclear_APAperm.png
Untracked: output/._qqplot_Nuclear_APAperm_4pc.png
Untracked: output/._qqplot_Total_APAperm.png
Untracked: output/._qqplot_Total_APAperm_4pc.png
Untracked: output/AverageDiffHeatmap.Nuclear.png
Untracked: output/AverageDiffHeatmap.Total.png
Untracked: output/GeneswithAPApotential.png
Untracked: output/GeneswithAPApotentialAllPAS.png
Untracked: output/PASlocation.png
Untracked: output/SignalSitePlot.png
Untracked: output/SignalSitePlotbyLoc.png
Untracked: output/dtPlots/
Untracked: output/fastqc/
Untracked: output/meanCorrelationPhenotypes.svg
Untracked: output/newnuc.png
Untracked: output/newtot.png
Untracked: output/oldnuc.png
Untracked: output/oldtot.png
Untracked: output/qqplot_Nuclear_APAperm.png
Untracked: output/qqplot_Nuclear_APAperm_4pc.png
Untracked: output/qqplot_Total_APAperm.png
Untracked: output/qqplot_Total_APAperm_4pc.png
Untracked: run_verifybam517N.err
Untracked: run_verifybam517N.out
Unstaged changes:
Modified: analysis/NuclearSpecIncludeNotTested.Rmd
Modified: analysis/PASdescriptiveplots.Rmd
Modified: analysis/Readdistagainstfeatures.Rmd
Modified: analysis/TSS.Rmd
Modified: analysis/apaQTLoverlap.Rmd
Modified: analysis/apabyeQTLstatus.Rmd
Modified: analysis/decayAndStability.Rmd
Modified: analysis/miRNAdisrupt.Rmd
Modified: analysis/nascenttranscription.Rmd
Modified: analysis/nucSpecinEQTLs.Rmd
Modified: analysis/overlapapaqtlsandeqtls.Rmd
Modified: analysis/pQTLexampleplot.Rmd
Modified: analysis/reads_graphs.Rmd
Modified: analysis/splicesitestrength.Rmd
Modified: analysis/version15bpfilter.Rmd
Modified: code/DistPAS2Sig.py
Modified: code/Script4NuclearQTLexamples.sh
Modified: code/Script4TotalQTLexamples.sh
Modified: code/apaQTLsnake.err
Modified: code/apaqtlfacetboxplots.R
Modified: code/environment.yaml
Modified: code/run_qtlFacetBoxplots.sh
Deleted: code/test.txt
Deleted: reads_graphs.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | b93e5d6 | brimittleman | 2020-03-23 | fix sup figs for revision |
| html | 4788c0e | brimittleman | 2020-02-18 | Build site. |
| Rmd | 8d8caba | brimittleman | 2020-02-18 | add lz |
| html | 00cd66c | brimittleman | 2019-09-10 | Build site. |
| Rmd | 5bcc43c | brimittleman | 2019-09-10 | test outlier removal |
| html | bec23da | brimittleman | 2019-09-06 | Build site. |
| Rmd | a2c35ac | brimittleman | 2019-09-06 | add correlation |
| html | 22541b3 | brimittleman | 2019-09-06 | Build site. |
| Rmd | 5976ed7 | brimittleman | 2019-09-06 | update post new filter |
| html | 27af5c8 | brimittleman | 2019-08-01 | Build site. |
| Rmd | e4d84f6 | brimittleman | 2019-08-01 | chamge pdf sizes for figure 3 |
| html | ab8482d | brimittleman | 2019-08-01 | Build site. |
| Rmd | 99c751e | brimittleman | 2019-08-01 | pdf for figure 3 |
| html | d73d818 | brimittleman | 2019-06-26 | Build site. |
| Rmd | c53925a | brimittleman | 2019-06-26 | add graph labels |
| html | 06de9df | brimittleman | 2019-06-26 | Build site. |
| Rmd | ec9c1d6 | brimittleman | 2019-06-26 | add direction concordance plots |
| html | ec8d7dc | brimittleman | 2019-06-26 | Build site. |
| Rmd | 52e46bc | brimittleman | 2019-06-26 | add example plot code |
| html | 0fae25e | brimittleman | 2019-06-20 | Build site. |
| Rmd | eb847c1 | brimittleman | 2019-06-20 | add analysis by pval |
| html | ca379ce | brimittleman | 2019-06-13 | Build site. |
| Rmd | 2fd2b27 | brimittleman | 2019-06-13 | fix bug |
| html | b907ac1 | brimittleman | 2019-06-12 | Build site. |
| Rmd | 178c5dc | brimittleman | 2019-06-12 | new geno |
| html | 6b164c8 | brimittleman | 2019-06-07 | Build site. |
| Rmd | b39620d | brimittleman | 2019-06-07 | add bonfor results |
| html | 458e494 | brimittleman | 2019-06-07 | Build site. |
| Rmd | 32091ee | brimittleman | 2019-06-07 | more prop explained to new analysis |
library(tidyverse)── Attaching packages ───────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ──────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(workflowr)This is workflowr version 1.6.0
Run ?workflowr for help getting startedlibrary(reshape2)
Attaching package: 'reshape2'The following object is masked from 'package:tidyr':
smithsI need to fix the explained_FDR10.sort.txt and unexplained_FDR10.sort.txt files because right now this file has multiple genes per snp.
python fixExandUnexeQTL.py ../data/Li_eQTLs/explained_FDR10.sort.txt ../data/Li_eQTLs/explained_FDR10.sort_FIXED.txt
python fixExandUnexeQTL.py ../data/Li_eQTLs/unexplained_FDR10.sort.txt ../data/Li_eQTLs/unexplained_FDR10.sort_FIXED.txtThere are 1195 explained and 814 unexplained eQTLs. I will next look at each of these in my apadata.
Convert nominal results to have snps rather than rsids:
python convertNominal2SNPLOC.py Total
python convertNominal2SNPLOC.py Nuclearmkdir ../data/overlapeQTL_try2
sbatch run_getapafromeQTL.sh
total
I can group the unexplained by gene and snp then I can ask if there is at least 1 significat peak for each of these.
I will use the bonforoni correction here and multiply the pvalue by the number of peaks in the gene:snp association.
nomnames=c("peakID", 'snp','dist', 'pval', 'slope')
totalapaUnexplained=read.table("../data/overlapeQTL_try2/apaTotal_unexplainedQTLs.txt", stringsAsFactors = F, col.names = nomnames)
totalapaUnexplained=totalapaUnexplained %>% separate(peakID, into=c("chr","start","end","geneID"), sep=":") %>% separate(geneID, into=c("gene", "loc", "strand", "PASnum"), sep="_") %>% group_by(gene, snp) %>% mutate(nPeaks=n(), adjPval=pval* nPeaks)%>% dplyr::slice(which.min(adjPval))
totalapaUnexplained_sig= totalapaUnexplained %>% filter(adjPval<.05)Look at distribution of these pvals:
ggplot(totalapaUnexplained, aes(x=adjPval)) + geom_histogram(bins=50)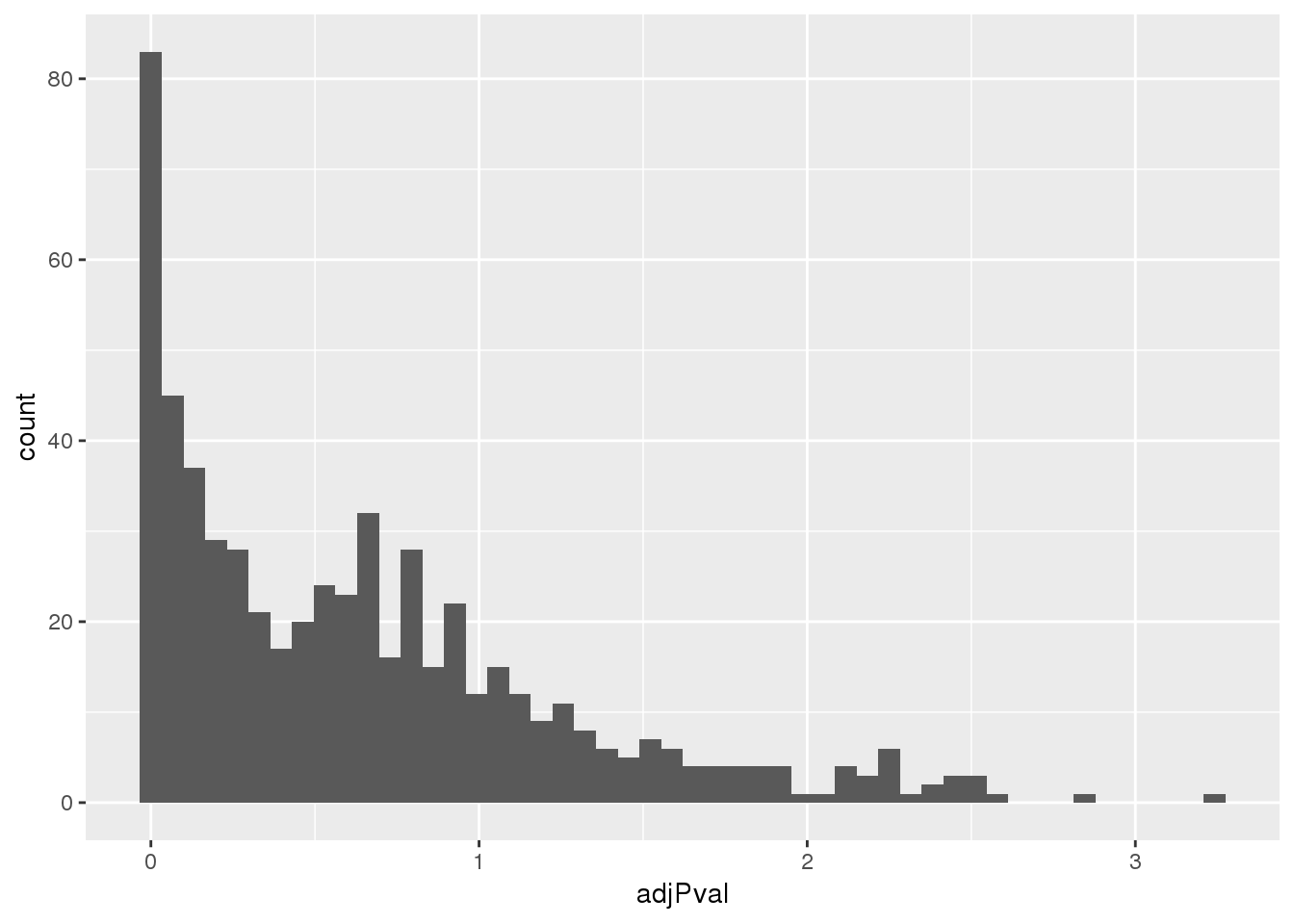
Proportion explained:
nrow(totalapaUnexplained_sig)/nrow(totalapaUnexplained)[1] 0.1678201Compare to explained eQTLS:
totalapaexplained=read.table("../data/overlapeQTL_try2/apaTotal_explainedQTLs.txt", stringsAsFactors = F, col.names = nomnames) %>% separate(peakID, into=c("chr","start","end","geneID"), sep=":") %>% separate(geneID, into=c("gene", "loc", "strand", "PASnum"), sep="_") %>% group_by(gene, snp) %>% mutate(nPeaks=n(), adjPval=pval* nPeaks) %>% dplyr::slice(which.min(adjPval))
totalapaexplained_sig= totalapaexplained %>% filter(adjPval<.05)
nrow(totalapaexplained_sig)/nrow(totalapaexplained)[1] 0.1248455difference of proportions:
prop.test(x=c(nrow(totalapaUnexplained_sig),nrow(totalapaexplained_sig)), n=c(nrow(totalapaUnexplained),nrow(totalapaexplained)))
2-sample test for equality of proportions with continuity
correction
data: c(nrow(totalapaUnexplained_sig), nrow(totalapaexplained_sig)) out of c(nrow(totalapaUnexplained), nrow(totalapaexplained))
X-squared = 4.7427, df = 1, p-value = 0.02942
alternative hypothesis: two.sided
95 percent confidence interval:
0.003452285 0.082496876
sample estimates:
prop 1 prop 2
0.1678201 0.1248455 ggplot(totalapaUnexplained_sig,aes(x=loc)) + geom_histogram(stat="count",aes(y=..count../sum(..count..))) + labs(y="Proportion", title = "Total apaQTLs explaining eQTLs")Warning: Ignoring unknown parameters: binwidth, bins, pad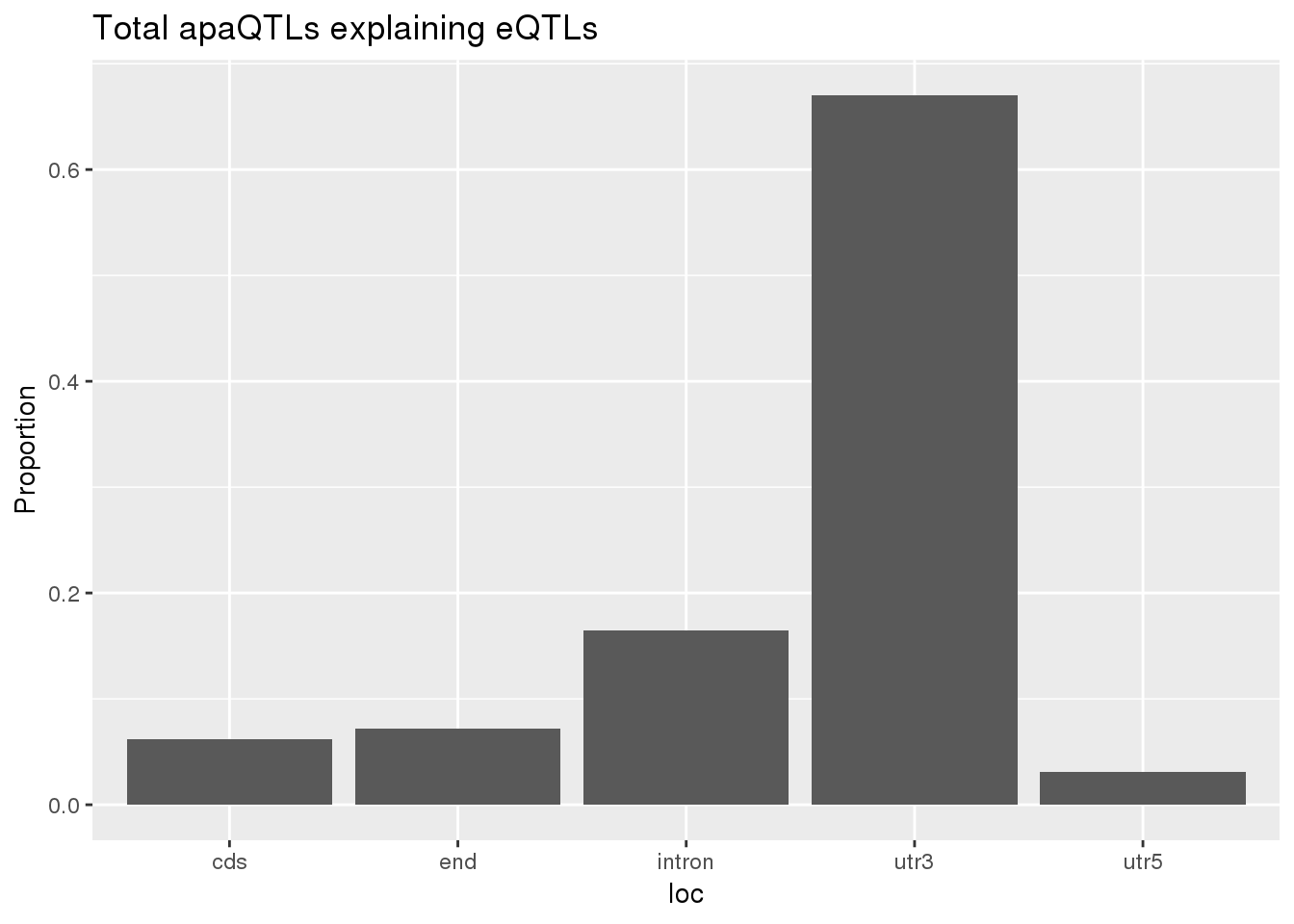
totalapaUnexplained_sig_loc= totalapaUnexplained_sig %>% group_by(loc) %>% summarise(nLocTotalUn=n()) %>% mutate(propTotalUn=nLocTotalUn/nrow(totalapaUnexplained_sig))
totalapaexplained_sig_loc= totalapaexplained_sig %>% group_by(loc) %>% summarise(nLocTotalEx=n()) %>% mutate(propTotalEx=nLocTotalEx/nrow(totalapaexplained_sig))
BothTotalLoc=totalapaUnexplained_sig_loc %>% full_join(totalapaexplained_sig_loc,by="loc") %>% replace_na(list(propTotalUn = 0, nLocTotalUn = 0,propTotalEx=0,nLocTotalEx=0 ))
BothTotalLoc# A tibble: 5 x 5
loc nLocTotalUn propTotalUn nLocTotalEx propTotalEx
<chr> <dbl> <dbl> <dbl> <dbl>
1 cds 6 0.0619 7 0.0693
2 end 7 0.0722 9 0.0891
3 intron 16 0.165 15 0.149
4 utr3 65 0.670 68 0.673
5 utr5 3 0.0309 2 0.0198nuclear
nuclearapaUnexplained=read.table("../data/overlapeQTL_try2/apaNuclear_unexplainedQTLs.txt", stringsAsFactors = F, col.names = nomnames) %>% separate(peakID, into=c("chr","start","end","geneID"), sep=":") %>% separate(geneID, into=c("gene", "loc", "strand", "PASnum"), sep="_") %>% group_by(gene, snp) %>% mutate(nPeaks=n(), adjPval=pval* nPeaks) %>% dplyr::slice(which.min(adjPval))
nuclearapaUnexplained_sig= nuclearapaUnexplained %>% filter(adjPval<.05)
nrow(nuclearapaUnexplained_sig)/nrow(nuclearapaUnexplained)[1] 0.1726496nuclearapaexplained=read.table("../data/overlapeQTL_try2/apaNuclear_explainedQTLs.txt", stringsAsFactors = F, col.names = nomnames) %>% separate(peakID, into=c("chr","start","end","geneID"), sep=":") %>% separate(geneID, into=c("gene", "loc", "strand", "PASnum"), sep="_") %>% group_by(gene, snp) %>% mutate(nPeaks=n(), adjPval=pval* nPeaks) %>% dplyr::slice(which.min(adjPval))
nuclearapaexplained_sig= nuclearapaexplained %>% filter(adjPval<.05)
nrow(nuclearapaexplained_sig)/nrow(nuclearapaexplained)[1] 0.1239264prop.test(x=c(nrow(nuclearapaUnexplained_sig),nrow(nuclearapaexplained_sig)), n=c(nrow(nuclearapaUnexplained),nrow(nuclearapaexplained)))
2-sample test for equality of proportions with continuity
correction
data: c(nrow(nuclearapaUnexplained_sig), nrow(nuclearapaexplained_sig)) out of c(nrow(nuclearapaUnexplained), nrow(nuclearapaexplained))
X-squared = 6.1593, df = 1, p-value = 0.01307
alternative hypothesis: two.sided
95 percent confidence interval:
0.009179856 0.088266529
sample estimates:
prop 1 prop 2
0.1726496 0.1239264 ggplot(nuclearapaUnexplained_sig,aes(x=loc)) + geom_histogram(stat="count",aes(y=..count../sum(..count..))) + labs(title = "Nuclear apaQTLs explaining eQTLs", y="Proportion")Warning: Ignoring unknown parameters: binwidth, bins, pad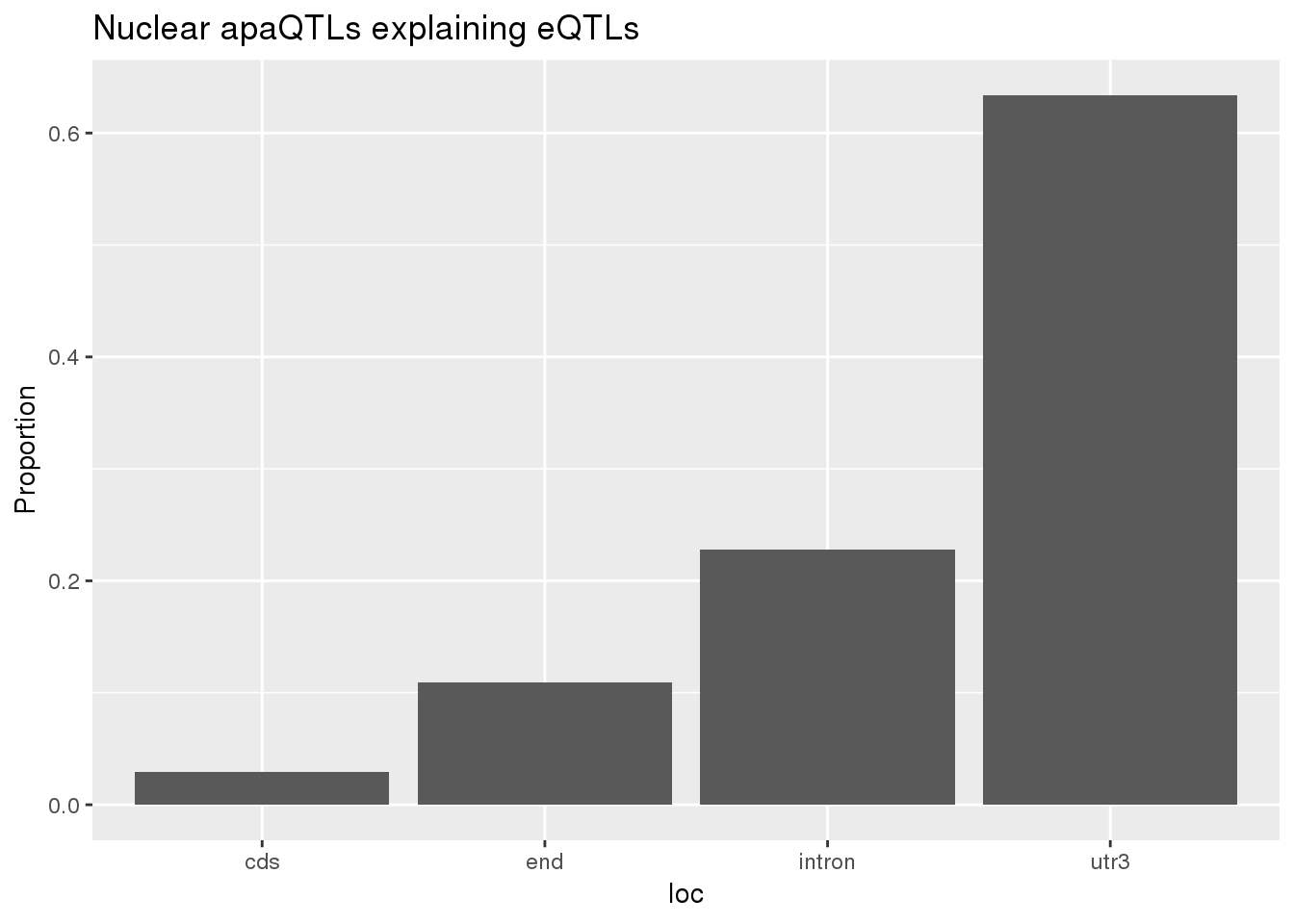
nuclearapaUnexplained_sig_loc= nuclearapaUnexplained_sig %>% group_by(loc) %>% summarise(nLocnuclearUn=n()) %>% mutate(propnuclearUn=nLocnuclearUn/nrow(nuclearapaUnexplained_sig))
nuclearapaexplained_sig_loc= nuclearapaexplained_sig %>% group_by(loc) %>% summarise(nLocnuclearEx=n()) %>% mutate(propnuclearEx=nLocnuclearEx/nrow(nuclearapaexplained_sig))
BothnuclearLoc=nuclearapaUnexplained_sig_loc %>% full_join(nuclearapaexplained_sig_loc,by="loc") %>% replace_na(list(propnuclearUn = 0, nLocnuclearUn = 0,propnuclearEx=0,nLocnuclearEx=0 ))
BothnuclearLoc# A tibble: 5 x 5
loc nLocnuclearUn propnuclearUn nLocnuclearEx propnuclearEx
<chr> <dbl> <dbl> <dbl> <dbl>
1 cds 3 0.0297 3 0.0297
2 end 11 0.109 12 0.119
3 intron 23 0.228 32 0.317
4 utr3 64 0.634 53 0.525
5 utr5 0 0 1 0.00990total v nuclear
prop.test(x=c(nrow(nuclearapaUnexplained_sig),nrow(totalapaUnexplained_sig)), n=c(nrow(nuclearapaUnexplained),nrow(totalapaUnexplained)))
2-sample test for equality of proportions with continuity
correction
data: c(nrow(nuclearapaUnexplained_sig), nrow(totalapaUnexplained_sig)) out of c(nrow(nuclearapaUnexplained), nrow(totalapaUnexplained))
X-squared = 0.019903, df = 1, p-value = 0.8878
alternative hypothesis: two.sided
95 percent confidence interval:
-0.04008930 0.04974831
sample estimates:
prop 1 prop 2
0.1726496 0.1678201 Differences in proportion by location
allLocProp=BothnuclearLoc %>% full_join(BothTotalLoc, by="loc") %>% select(loc,propnuclearUn,propnuclearEx,propTotalUn,propTotalEx )
allLocPropmelt= melt(allLocProp, id.vars = "loc") %>% mutate(Fraction=ifelse(grepl("Total", variable), "Total", "Nuclear"),eQTL=ifelse(grepl("Un", variable), "Unexplained", "Explained"))
ggplot(allLocPropmelt,aes(x=loc, fill=eQTL, y=value)) + geom_histogram(stat="identity", position = "dodge") + facet_grid(~Fraction)+ labs(y="Proportion of PAS", title="apaQTLs overlaping eQTLs by PAS location") + scale_fill_manual(values=c("orange", "blue"))Warning: Ignoring unknown parameters: binwidth, bins, pad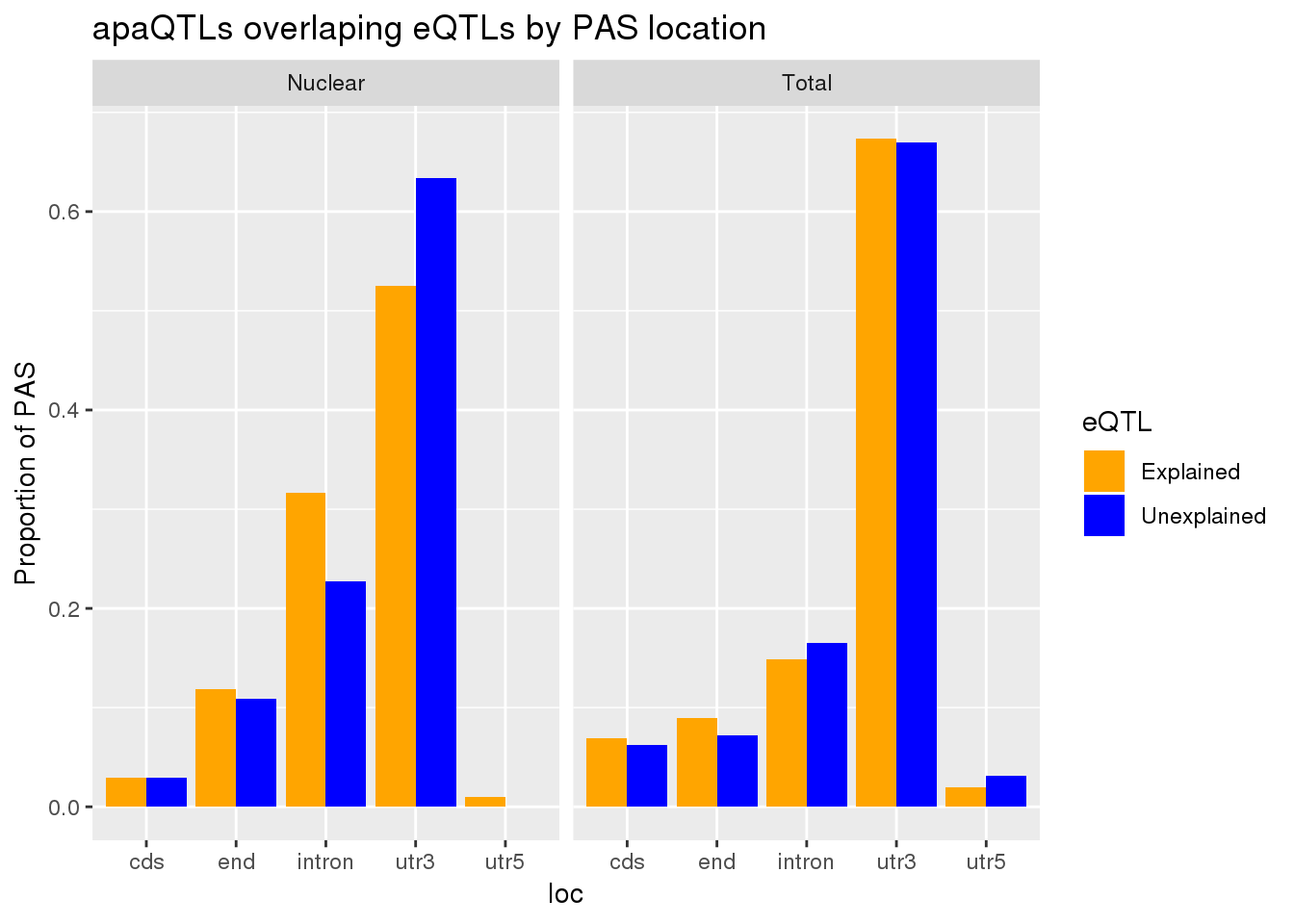
This is a very stringent test. A less stringent way to get an upper bound would be to make an informed decision about which peak to use. This will make it so I am only testing one PAS per gene.
Vary the pvalue cuttoff
To test if .05 is a good cuttoff for this analysis I will create a function that computes the overlap at different cutoffs. I will go from .01 to .5 by .05
totalapaUnexplained totalapaexplained
nuclearapaUnexplained nuclearapaexplained
prop_overlap=function(status, fraction, cutoff){
if (fraction=="Total"){
if (status=="Explained"){
file=totalapaexplained
sig=file %>% filter(adjPval<=cutoff)
proportion=round(nrow(sig)/nrow(file),digits=2)
}else {
file=totalapaUnexplained
sig=file %>% filter(adjPval<=cutoff)
proportion=round(nrow(sig)/nrow(file),digits=2)
}
} else{
if (status=="Explained"){
file=nuclearapaexplained
sig=file %>% filter(adjPval<=cutoff)
proportion=round(nrow(sig)/nrow(file),digits=2)
}else {
file=nuclearapaUnexplained
sig=file %>% filter(adjPval<=cutoff)
proportion=round(nrow(sig)/nrow(file),digits=2)
}
}
return(proportion)
}cutoffs=c(0.001,0.01,0.02,0.03,0.04,0.05,0.1,0.2,0.3,0.4,0.5)
TotalExplained_Proportions=c()
for(i in cutoffs){
TotalExplained_Proportions=c( TotalExplained_Proportions, prop_overlap("Explained", "Total", i))
}
TotalExplained_ProportionsDF=as.data.frame(cbind(cutoffs,Prop=TotalExplained_Proportions, Status=rep("Explained", 11), Fraction=rep("Total", 11)))
TotalUnexplained_Proportions=c()
for(i in cutoffs){
TotalUnexplained_Proportions=c(TotalUnexplained_Proportions, prop_overlap("Unexplained", "Total", i))
}
TotalUnexplained_ProportionsDF=as.data.frame(cbind(cutoffs,Prop=TotalUnexplained_Proportions, Status=rep("Unexplained", 11), Fraction=rep("Total", 11)))
NuclearExplained_Proportions=c()
for(i in cutoffs){
NuclearExplained_Proportions=c( NuclearExplained_Proportions, prop_overlap("Explained", "Nuclear", i))
}
NuclearExplained_ProportionsDF=as.data.frame(cbind(cutoffs,Prop=NuclearExplained_Proportions, Status=rep("Explained", 11), Fraction=rep("Nuclear", 11)))
NuclearUnexplained_Proportions=c()
for(i in cutoffs){
NuclearUnexplained_Proportions=c( NuclearUnexplained_Proportions, prop_overlap("Unexplained", "Nuclear", i))
}
NuclearUnexplained_ProportionsDF=as.data.frame(cbind(cutoffs,Prop=NuclearUnexplained_Proportions, Status=rep("Unexplained", 11), Fraction=rep("Nuclear", 11)))
AllPropDF=bind_rows(TotalExplained_ProportionsDF,TotalUnexplained_ProportionsDF,NuclearExplained_ProportionsDF,NuclearUnexplained_ProportionsDF)Warning in bind_rows_(x, .id): Unequal factor levels: coercing to characterWarning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vectorWarning in bind_rows_(x, .id): Unequal factor levels: coercing to characterWarning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vectorWarning in bind_rows_(x, .id): Unequal factor levels: coercing to characterWarning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vector
Warning in bind_rows_(x, .id): binding character and factor vector,
coercing into character vectorAllPropDF$Prop=as.numeric(AllPropDF$Prop)Plot this:
ggplot(AllPropDF, aes(x=cutoffs, y=Prop, fill=Status)) + geom_bar(position = "dodge", stat="identity") + facet_grid(~Fraction) + labs(title="Proportion of eQTLs explained by apaQTLs", y="Proportion", "P-Value cut off") + scale_fill_manual(values=c("orange", "blue")) + theme(axis.text.x = element_text(angle = 90, hjust = .5))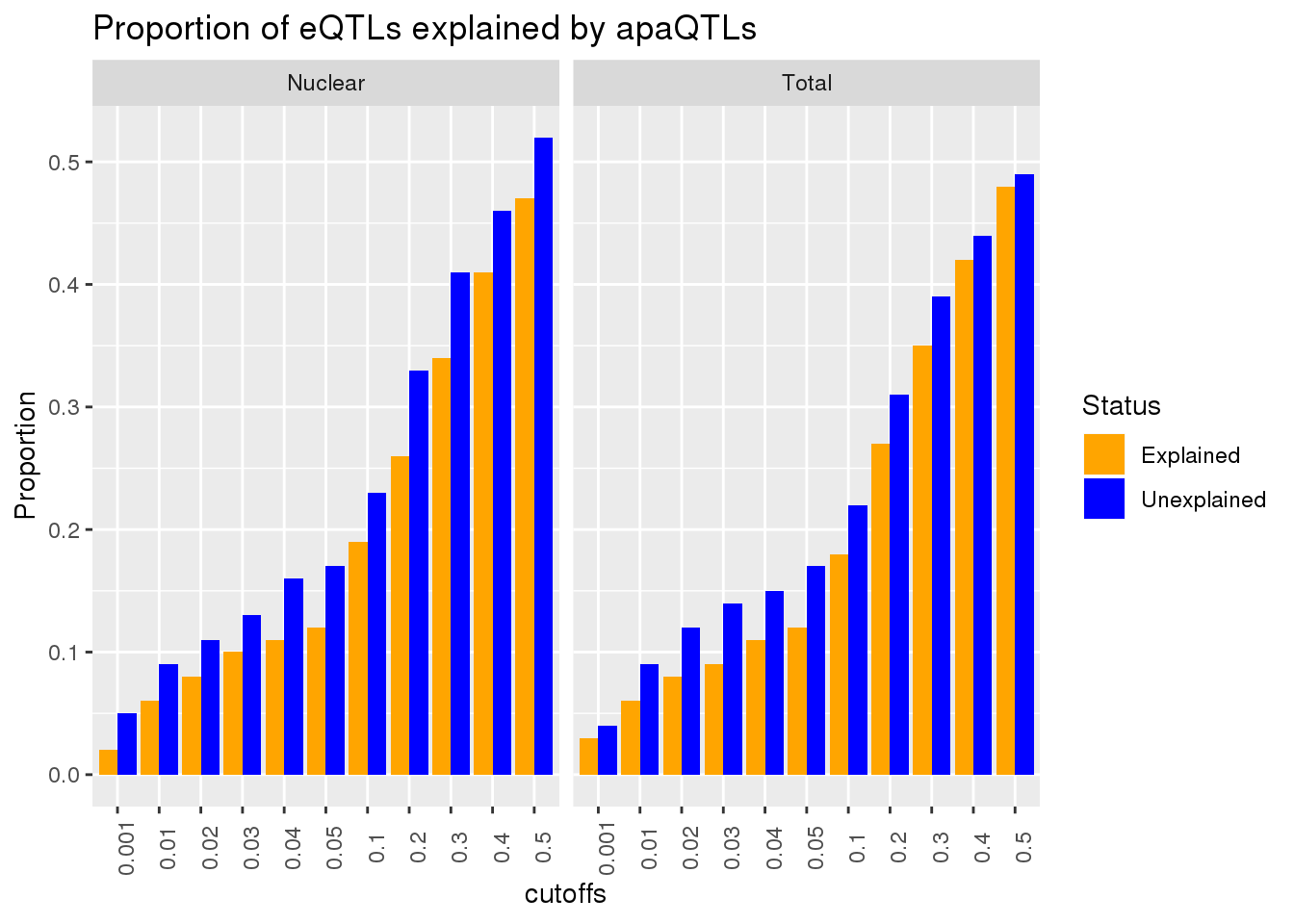
| Version | Author | Date |
|---|---|---|
| 22541b3 | brimittleman | 2019-09-06 |
Concordance of directions for intronic pas usage and eQTL
I want to look at the intronic pas and the eQTLs. To do this I want to look at a correaltion of effect sizes for the eQTLs and and intronic PAS.
The eQTL information is in ../data/molQTLs/fastqtl_qqnorm_RNAseq_phase2.fixed.nominal.AllNomRes.GeneName.txt. I need to converte the RSID into snp loc.
python eQTL_switch2snploc.pyprepare eQTL:
eQTLeffect=read.table("../data/molQTLs/fastqtl_qqnorm_RNAseq_phase2.fixed.nominal.AllNomRes.GeneName_snploc.txt", stringsAsFactors = F, col.names = c("gene","snp","dist", "pval", "eQTL_es")) %>% select(gene, snp, eQTL_es)total:
#totalunex_all=read.table("../data/overlapeQTL_try2/apaTotal_unexplainedQTLs.txt", stringsAsFactors = F, col.names = nomnames) %>% separate(peakID, into=c("chr","start","end","geneID"), sep=":") %>% separate(geneID, into=c("gene", "loc", "strand", "PASnum"), sep="_")
#totalex_all=read.table("../data/overlapeQTL_try2/apaTotal_explainedQTLs.txt", stringsAsFactors = F, col.names = nomnames) %>% separate(peakID, into=c("chr","start","end","geneID"), sep=":") %>% separate(geneID, into=c("gene", "loc", "strand", "PASnum"), sep="_")
alleQTLS_total=bind_rows(totalapaUnexplained, totalapaexplained) %>% filter(loc=="intron") %>% inner_join(eQTLeffect, by=c("gene","snp"))
ggplot(alleQTLS_total,aes(x=eQTL_es, y=slope)) + geom_point() + geom_smooth(method = "lm") +geom_text(y=-1, x=-1.5, label="slope: -0.22 p-value: 0.00002, r2=0.08") + labs(title="Total apa effect sizes vs eQTL eqtl effect sizes", y="Total apaQTL effect size",x="eQTL effect size")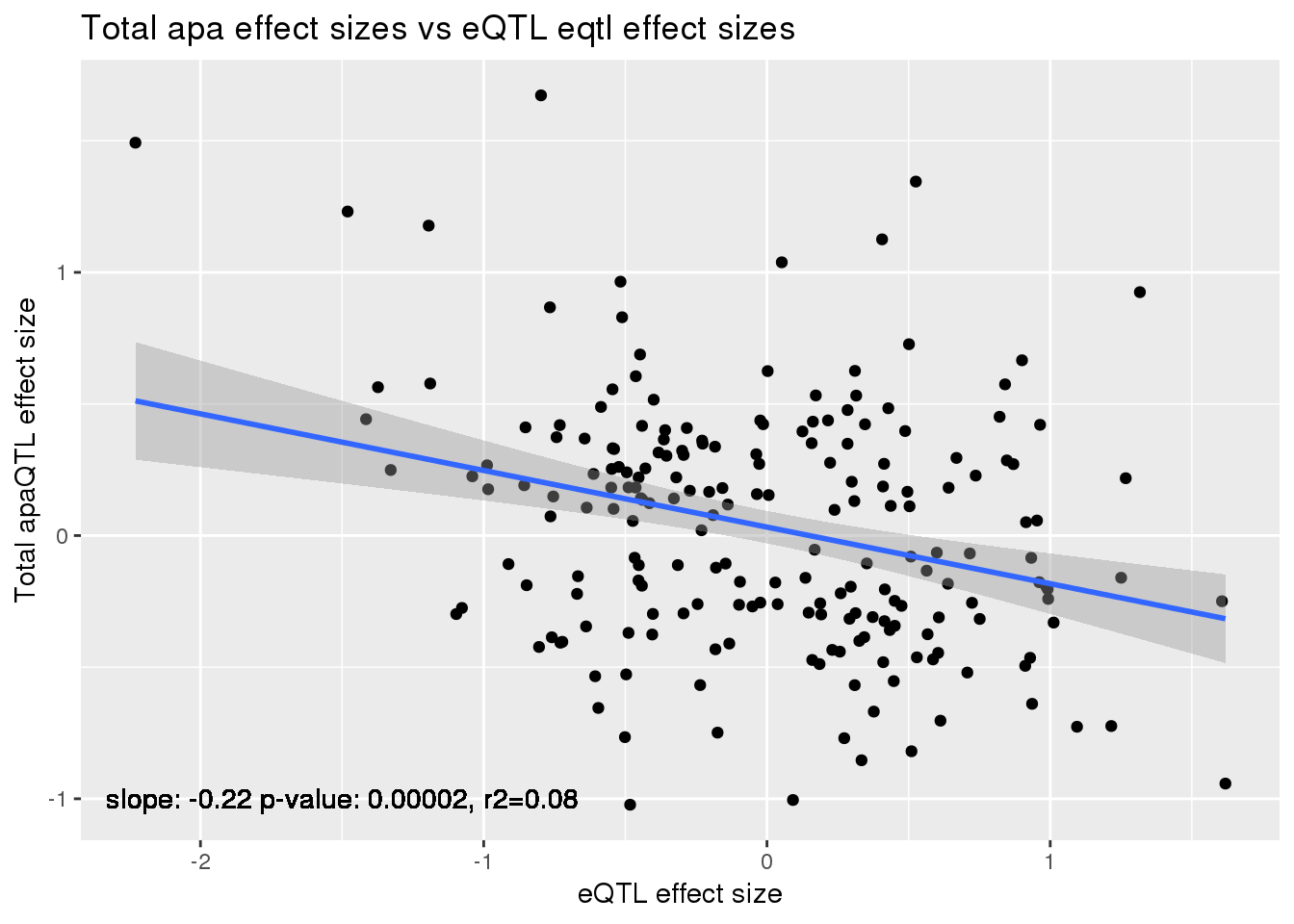
| Version | Author | Date |
|---|---|---|
| 22541b3 | brimittleman | 2019-09-06 |
summary(lm(alleQTLS_total$slope ~alleQTLS_total$eQTL_es))
Call:
lm(formula = alleQTLS_total$slope ~ alleQTLS_total$eQTL_es)
Residuals:
Min 1Q Median 3Q Max
-1.15866 -0.31339 -0.00043 0.26661 1.46869
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 0.03214 0.03132 1.026 0.306
alleQTLS_total$eQTL_es -0.21510 0.04901 -4.389 1.83e-05 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.4474 on 202 degrees of freedom
Multiple R-squared: 0.08707, Adjusted R-squared: 0.08255
F-statistic: 19.27 on 1 and 202 DF, p-value: 1.833e-05cor.test(alleQTLS_total$slope ,alleQTLS_total$eQTL_es, alternative="less")
Pearson's product-moment correlation
data: alleQTLS_total$slope and alleQTLS_total$eQTL_es
t = -4.3892, df = 202, p-value = 9.163e-06
alternative hypothesis: true correlation is less than 0
95 percent confidence interval:
-1.000000 -0.185907
sample estimates:
cor
-0.2950724 Nuclear:
alleQTLS_nuclear=bind_rows(nuclearapaUnexplained,nuclearapaexplained) %>% filter(loc=="intron") %>% inner_join(eQTLeffect, by=c("gene","snp"))
ggplot(alleQTLS_nuclear,aes(x=eQTL_es, y=slope)) + geom_point() + geom_smooth(method = "lm") +geom_text(y=1.5, x=-1, label="slope: -0.20 p-value: 9.0 * 10 ^ -9, r2=0.08") + labs(title="", y="apaQTL effect size",x="eQTL effect size")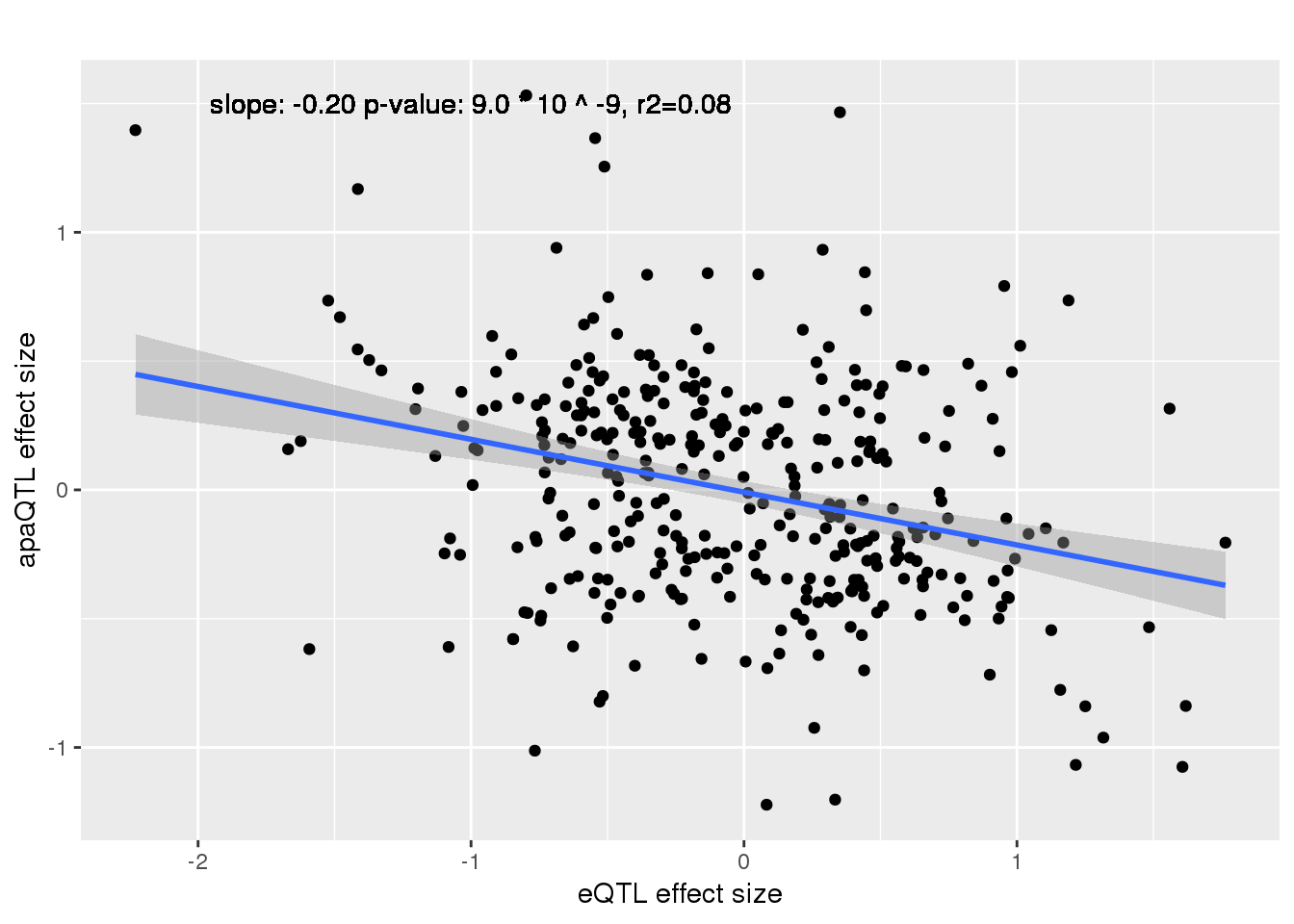
summary(lm(alleQTLS_nuclear$slope ~alleQTLS_nuclear$eQTL_es))
Call:
lm(formula = alleQTLS_nuclear$slope ~ alleQTLS_nuclear$eQTL_es)
Residuals:
Min 1Q Median 3Q Max
-1.19658 -0.29003 -0.00934 0.26184 1.54707
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -0.008894 0.022167 -0.401 0.688
alleQTLS_nuclear$eQTL_es -0.205079 0.034819 -5.890 8.97e-09 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.418 on 355 degrees of freedom
Multiple R-squared: 0.08902, Adjusted R-squared: 0.08646
F-statistic: 34.69 on 1 and 355 DF, p-value: 8.97e-09cor.test(alleQTLS_nuclear$slope, alleQTLS_nuclear$eQTL_es, alternative="less")
Pearson's product-moment correlation
data: alleQTLS_nuclear$slope and alleQTLS_nuclear$eQTL_es
t = -5.8899, df = 355, p-value = 4.485e-09
alternative hypothesis: true correlation is less than 0
95 percent confidence interval:
-1.0000000 -0.2168049
sample estimates:
cor
-0.2983651 remove outlier and see if it holds:
alleQTLS_nuclear_noOut=alleQTLS_nuclear %>% filter(eQTL_es > -2)
ggplot(alleQTLS_nuclear_noOut,aes(x=eQTL_es, y=slope)) + geom_point() + geom_smooth(method = "lm") + labs(title="", y="apaQTL effect size",x="eQTL effect size")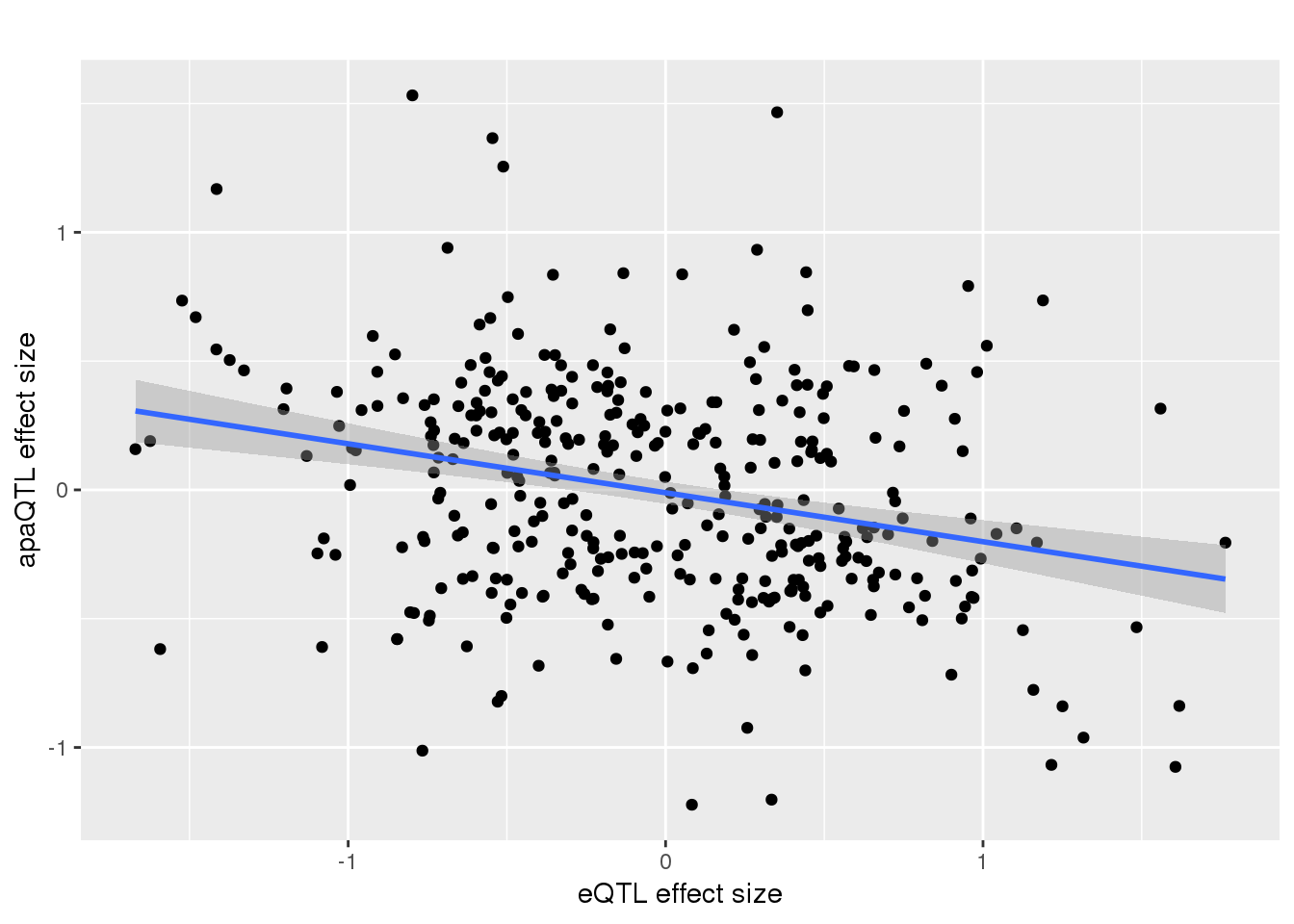
summary(lm(alleQTLS_nuclear_noOut$slope ~alleQTLS_nuclear_noOut$eQTL_es))
Call:
lm(formula = alleQTLS_nuclear_noOut$slope ~ alleQTLS_nuclear_noOut$eQTL_es)
Residuals:
Min 1Q Median 3Q Max
-1.19565 -0.29112 -0.00921 0.25549 1.54399
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -0.01106 0.02205 -0.502 0.616
alleQTLS_nuclear_noOut$eQTL_es -0.19013 0.03520 -5.402 1.21e-07 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.4155 on 354 degrees of freedom
Multiple R-squared: 0.07615, Adjusted R-squared: 0.07354
F-statistic: 29.18 on 1 and 354 DF, p-value: 1.213e-07Examples for overlap:
unexplained_snps=read.table("../data/Li_eQTLs/unexplained_FDR10.sort_FIXED.txt", col.names = c("chr", "loc", "gene"),stringsAsFactors = F)
totQTL=read.table("../data/apaQTLs/Total_apaQTLs4pc_5fdr.bed", header = T, stringsAsFactors = F, col.names = c("chr", "bedstart","loc","ID", "score", "strand"))
nucQTL=read.table("../data/apaQTLs/Nuclear_apaQTLs4pc_5fdr.bed", stringsAsFactors = F, header = T, col.names = c("chr", "bedstart","loc","ID", "score", "strand"))Overlap:
totQTL_unex=totQTL %>% inner_join(unexplained_snps, by=c("chr", "loc"))
nucQTL_unex=nucQTL %>% inner_join(unexplained_snps, by=c("chr", "loc"))totQTL_unex chr bedstart loc ID score strand
1 10 124693586 124693587 C10orf88:peak19682:intron 0.829354 .
2 19 57706377 57706378 ZNF264:peak67214:intron -0.765818 .
3 20 1350708 1350709 FKBP1A:peak79304:utr3 -0.569411 .
4 2 197855151 197855152 ANKRD44:peak76705:utr3 0.464009 .
5 2 197855151 197855152 ANKRD44:peak76708:intron -1.022620 .
6 6 44275010 44275011 AARS2:peak113590:utr3 0.968958 .
7 7 6497500 6497501 KDELR2:peak118586:utr3 1.003000 .
8 7 6497500 6497501 KDELR2:peak118588:utr3 -1.032740 .
gene
1 C10orf88
2 ZNF264
3 FKBP1A
4 ANKRD44
5 ANKRD44
6 AARS2
7 KDELR2
8 KDELR2nucQTL_unex chr bedstart loc ID score strand
1 10 124693586 124693587 C10orf88:peak19682:intron 1.255120 .
2 19 57706377 57706378 ZNF264:peak67214:intron -0.496966 .
3 4 44702719 44702720 GUF1:peak97168:utr3 0.882583 .
4 4 44702719 44702720 GUF1:peak97169:utr3 -1.377620 .
gene
1 C10orf88
2 ZNF264
3 GNPDA2
4 GNPDA2Make a plot for KDELR2 7:6497501
genohead=as.data.frame(read.table("../data/ExampleQTLPlots/genotypeHeader.txt", stringsAsFactors = F, header = F)[,10:128] %>% t())
colnames(genohead)=c("header")
genotype=as.data.frame(read.table("../data/ExampleQTLPlots/KDELR2_TotalPeaksGenotype.txt", stringsAsFactors = F, header = F) [,10:128] %>% t())
full_geno=bind_cols(Ind=genohead$header, dose=genotype$V1) %>% mutate(numdose=round(dose), genotype=ifelse(numdose==0, "TT", ifelse(numdose==1, "TG", "GG")))
RNAhead=as.data.frame(read.table("../data/molPhenos/RNAhead.txt", stringsAsFactors = F, header = F)[,5:73] %>% t())
RNApheno=as.data.frame(read.table("../data/molPhenos/RNA_KDELr2.txt", stringsAsFactors = F, header = F) [,5:73] %>% t())
full_pheno=bind_cols(Ind=RNAhead$V1, Expression=RNApheno$V1)
allRNA=full_geno %>% inner_join(full_pheno, by="Ind")
allRNA$genotype=as.factor(allRNA$genotype)Ref,T Alt= G
ggplot(allRNA, aes(x=genotype, y=Expression,group=genotype, fill=genotype)) + geom_boxplot() + geom_jitter()+scale_fill_brewer(palette = "YlOrRd") + labs(title="Unexplained eQTL: KDELR2 - rs6962012")Make locus zoom
mkdir ../data/locusZoompeak119699 KDELR2 ENSG00000136240.5
grep peak119699 ../data/apaQTLNominal_4pc/APApeak_Phenotype_GeneLocAnno.Total.5perc.fc.gz.qqnorm_AllChrom.txt > ../data/locusZoom/TotalAPA.peak119699.KDELR2.nomNuc.txt
grep ENSG00000136240.5 ../data/molQTLs/fastqtl_qqnorm_RNAseq_phase2.fixed.nominal.AllNomRes.txt > ../data/locusZoom/RNA.KDELR2.txt
APATotal_KDELR2_LZ=read.table("../data/locusZoom/TotalAPA.peak119699.KDELR2.nomNuc.txt", stringsAsFactors = F, col.names = c("PeakID", "SNP", "Dist", "P","slope")) %>% select( SNP, P)
write.table(APATotal_KDELR2_LZ,"../data/locusZoom/apaTotalKDELR2_LZ.txt", col.names = T, row.names = F, quote = F)
RNA_KDELR2_LZ=read.table("../data/locusZoom/RNA.KDELR2.txt", stringsAsFactors = F, col.names = c("PeakID", "SNP", "Dist", "P","slope")) %>% select( SNP, P)
write.table(RNA_KDELR2_LZ,"../data/locusZoom/RNAKDELR2_LZ.txt", col.names = T, row.names = F, quote = F)Use locuszoom.org
locus zoom plot for C10ofr88 variant in nuclear:
peak19682
grep peak19682 ../data/apaQTLNominal_4pc/APApeak_Phenotype_GeneLocAnno.Nuclear.5perc.fc.gz.qqnorm_AllChrom.txt > ../data/locusZoom/NuclearAPA.peak19882.C10ofr88.nomNuc.txt
grep ENSG00000119965 ../data/molQTLs/fastqtl_qqnorm_RNAseq_phase2.fixed.nominal.AllNomRes.txt > ../data/locusZoom/RNA.C10ofr88.txt
APATNuclear_orf_LZ=read.table("../data/locusZoom/NuclearAPA.peak19882.C10ofr88.nomNuc.txt", stringsAsFactors = F, col.names = c("PeakID", "SNP", "Dist", "P","slope")) %>% select( SNP, P)
write.table(APATNuclear_orf_LZ,"../data/locusZoom/apaNuclearC10orf88_LZ.txt", col.names = T, row.names = F, quote = F)
RNA_orf_LZ=read.table("../data/locusZoom/RNA.C10ofr88.txt", stringsAsFactors = F, col.names = c("PeakID", "SNP", "Dist", "P","slope")) %>% select( SNP, P)
write.table(RNA_orf_LZ,"../data/locusZoom/RNAC10orf88_LZ.txt", col.names = T, row.names = F, quote = F)Locus zoom for all of the examples
module load R
module load plink
module load htslib
mkdir ../data/eQTL_LZ
mkdir ../data/eQTL_LZ/NuclearAssoc/
mkdir ../data/eQTL_LZ/RNAAssoc/I need to extract the PAS and genes from the nominal files. Do this for nuclear. The below dataframes come from looking at the original eQTL snp in the apa data. I choose the more sig PAS per gene. I will use these for this as well.
I need to get the RSIDs for these
RSID=read.table("/project2/gilad/briana/li_genotypes/RSID2snploc.txt",header = T, stringsAsFactors = F)
AllNuclear_sig= nuclearapaexplained_sig %>% bind_rows(nuclearapaUnexplained_sig) %>% inner_join(RSID, by="snp") %>% ungroup() %>% select(gene, PASnum, RSID)
write.table(AllNuclear_sig,"../data/eQTL_LZ/PasGENEsnpstoUse.txt", col.names = F, row.names = F, quote = F)sbatch ExtractPAS4eQTLsLZ.sh
cd ../data/eQTL_LZ/NuclearAssoc
sbatch CreateAPALZeQTLs.sh
sbatch extractGeneLZfileseQTLs.sh
cd ../data/eQTL_LZ/RNAAssoc
sbatch CreateRNALZforeQTLs.sh
Looks like a lot of these do. I can use the snps for explained vs unexplained to copy them to seperate files.
explainedRS=nuclearapaexplained_sig %>% inner_join(RSID, by="snp") %>% ungroup() %>% select(RSID)
write.table(explainedRS, "../data/eQTL_LZ/explainedRS.txt", col.names = F, row.names = F, quote = F)
UnexplainedRS=nuclearapaUnexplained_sig %>% inner_join(RSID, by="snp") %>% ungroup() %>% select(RSID)
write.table(UnexplainedRS, "../data/eQTL_LZ/UnexplainedRS.txt", col.names = F, row.names = F, quote = F)I need a way to use these lists to move the correct plots to seperate places
SWITCH DIR
mkdir UnexplainedeQTLs
mkdir ExplainedeQTLsI can do this in bash.
cat UnexplainedRS.txt | while read line
do
read -a strarr <<< $line
cp NuclearAssoc/200217_${line}/*.pdf UnexplainedeQTLs
done
cat explainedRS.txt | while read line
do
read -a strarr <<< $line
cp NuclearAssoc/200217_${line}/*.pdf ExplainedeQTLs
done
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] reshape2_1.4.3 workflowr_1.6.0 forcats_0.3.0 stringr_1.3.1
[5] dplyr_0.8.0.1 purrr_0.3.2 readr_1.3.1 tidyr_0.8.3
[9] tibble_2.1.1 ggplot2_3.1.1 tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 haven_1.1.2 lattice_0.20-38 colorspace_1.3-2
[5] generics_0.0.2 htmltools_0.3.6 yaml_2.2.0 utf8_1.1.4
[9] rlang_0.4.0 later_0.7.5 pillar_1.3.1 glue_1.3.0
[13] withr_2.1.2 modelr_0.1.2 readxl_1.1.0 plyr_1.8.4
[17] munsell_0.5.0 gtable_0.2.0 cellranger_1.1.0 rvest_0.3.2
[21] evaluate_0.12 labeling_0.3 knitr_1.20 httpuv_1.4.5
[25] fansi_0.4.0 broom_0.5.1 Rcpp_1.0.2 promises_1.0.1
[29] scales_1.0.0 backports_1.1.2 jsonlite_1.6 fs_1.3.1
[33] hms_0.4.2 digest_0.6.18 stringi_1.2.4 grid_3.5.1
[37] rprojroot_1.3-2 cli_1.1.0 tools_3.5.1 magrittr_1.5
[41] lazyeval_0.2.1 crayon_1.3.4 whisker_0.3-2 pkgconfig_2.0.2
[45] xml2_1.2.0 lubridate_1.7.4 assertthat_0.2.0 rmarkdown_1.10
[49] httr_1.3.1 rstudioapi_0.10 R6_2.3.0 nlme_3.1-137
[53] git2r_0.26.1 compiler_3.5.1