Overlap APAqtls with CTCF qtls
Briana Mittleman
3/19/2019
Last updated: 2019-03-20
Checks: 6 0
Knit directory: threeprimeseq/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.2.0). The Report tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(12345) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/.DS_Store
Ignored: data/perm_QTL_trans_noMP_5percov/
Ignored: output/.DS_Store
Untracked files:
Untracked: KalistoAbundance18486.txt
Untracked: analysis/4suDataIGV.Rmd
Untracked: analysis/DirectionapaQTL.Rmd
Untracked: analysis/EvaleQTLs.Rmd
Untracked: analysis/YL_QTL_test.Rmd
Untracked: analysis/groSeqAnalysis.Rmd
Untracked: analysis/ncbiRefSeq_sm.sort.mRNA.bed
Untracked: analysis/snake.config.notes.Rmd
Untracked: analysis/verifyBAM.Rmd
Untracked: analysis/verifybam_dubs.Rmd
Untracked: code/PeaksToCoverPerReads.py
Untracked: code/strober_pc_pve_heatmap_func.R
Untracked: data/18486.genecov.txt
Untracked: data/APApeaksYL.total.inbrain.bed
Untracked: data/AllPeak_counts/
Untracked: data/ApaQTLs/
Untracked: data/ApaQTLs_otherPhen/
Untracked: data/CTCF/
Untracked: data/ChromHmmOverlap/
Untracked: data/DistTXN2Peak_genelocAnno/
Untracked: data/EmpiricalDists/
Untracked: data/FeatureoverlapPeaks/
Untracked: data/GM12878.chromHMM.bed
Untracked: data/GM12878.chromHMM.txt
Untracked: data/GWAS_overlap/
Untracked: data/LianoglouLCL/
Untracked: data/LocusZoom/
Untracked: data/LocusZoom_Unexp/
Untracked: data/LocusZoom_proc/
Untracked: data/MatchedSnps/
Untracked: data/NucSpecQTL/
Untracked: data/NuclearApaQTLs.txt
Untracked: data/PeakCounts/
Untracked: data/PeakCounts_noMP_5perc/
Untracked: data/PeakCounts_noMP_genelocanno/
Untracked: data/PeakUsage/
Untracked: data/PeakUsage_noMP/
Untracked: data/PeakUsage_noMP_GeneLocAnno/
Untracked: data/PeaksUsed/
Untracked: data/PeaksUsed_noMP_5percCov/
Untracked: data/PolyA_DB/
Untracked: data/QTL_overlap/
Untracked: data/RNAdecay/
Untracked: data/RNAkalisto/
Untracked: data/RefSeq_annotations/
Untracked: data/Replicates_usage/
Untracked: data/Signal_Loc/
Untracked: data/TotalApaQTLs.txt
Untracked: data/Totalpeaks_filtered_clean.bed
Untracked: data/UnderstandPeaksQC/
Untracked: data/WASP_STAT/
Untracked: data/YL-SP-18486-T-combined-genecov.txt
Untracked: data/YL-SP-18486-T_S9_R1_001-genecov.txt
Untracked: data/YL_QTL_test/
Untracked: data/apaExamp/
Untracked: data/apaExamp_proc/
Untracked: data/apaQTL_examp_noMP/
Untracked: data/bedgraph_peaks/
Untracked: data/bin200.5.T.nuccov.bed
Untracked: data/bin200.Anuccov.bed
Untracked: data/bin200.nuccov.bed
Untracked: data/clean_peaks/
Untracked: data/comb_map_stats.csv
Untracked: data/comb_map_stats.xlsx
Untracked: data/comb_map_stats_39ind.csv
Untracked: data/combined_reads_mapped_three_prime_seq.csv
Untracked: data/diff_iso_GeneLocAnno/
Untracked: data/diff_iso_proc/
Untracked: data/diff_iso_trans/
Untracked: data/eQTL_inAPA/
Untracked: data/eQTLs_Lietal/
Untracked: data/ensemble_to_genename.txt
Untracked: data/example_gene_peakQuant/
Untracked: data/explainProtVar/
Untracked: data/filtPeakOppstrand_cov_noMP_GeneLocAnno_5perc/
Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.bed
Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.noties.bed
Untracked: data/first50lines_closest.txt
Untracked: data/gencov.test.csv
Untracked: data/gencov.test.txt
Untracked: data/gencov_zero.test.csv
Untracked: data/gencov_zero.test.txt
Untracked: data/gene_cov/
Untracked: data/joined
Untracked: data/leafcutter/
Untracked: data/merged_combined_YL-SP-threeprimeseq.bg
Untracked: data/molPheno_noMP/
Untracked: data/mol_overlap/
Untracked: data/mol_pheno/
Untracked: data/nom_QTL/
Untracked: data/nom_QTL_opp/
Untracked: data/nom_QTL_trans/
Untracked: data/nuc6up/
Untracked: data/nuc_10up/
Untracked: data/other_qtls/
Untracked: data/pQTL_inAPA/
Untracked: data/pQTL_otherphen/
Untracked: data/pacbio_cov/
Untracked: data/peakPerRefSeqGene/
Untracked: data/peaks4DT/
Untracked: data/perm_QTL/
Untracked: data/perm_QTL_GeneLocAnno_noMP_5percov/
Untracked: data/perm_QTL_GeneLocAnno_noMP_5percov_3UTR/
Untracked: data/perm_QTL_diffWindow/
Untracked: data/perm_QTL_opp/
Untracked: data/perm_QTL_trans/
Untracked: data/perm_QTL_trans_filt/
Untracked: data/protAndAPAAndExplmRes.Rda
Untracked: data/protAndAPAlmRes.Rda
Untracked: data/protAndExpressionlmRes.Rda
Untracked: data/reads_mapped_three_prime_seq.csv
Untracked: data/smash.cov.results.bed
Untracked: data/smash.cov.results.csv
Untracked: data/smash.cov.results.txt
Untracked: data/smash_testregion/
Untracked: data/ssFC200.cov.bed
Untracked: data/temp.file1
Untracked: data/temp.file2
Untracked: data/temp.gencov.test.txt
Untracked: data/temp.gencov_zero.test.txt
Untracked: data/threePrimeSeqMetaData.csv
Untracked: data/threePrimeSeqMetaData55Ind.txt
Untracked: data/threePrimeSeqMetaData55Ind.xlsx
Untracked: data/threePrimeSeqMetaData55Ind_noDup.txt
Untracked: data/threePrimeSeqMetaData55Ind_noDup.xlsx
Untracked: data/threePrimeSeqMetaData55Ind_noDup_WASPMAP.txt
Untracked: data/threePrimeSeqMetaData55Ind_noDup_WASPMAP.xlsx
Untracked: manuscript/
Untracked: output/LZ/
Untracked: output/deeptools_plots/
Untracked: output/picard/
Untracked: output/plots/
Untracked: output/qual.fig2.pdf
Unstaged changes:
Modified: analysis/28ind.peak.explore.Rmd
Modified: analysis/CompareLianoglouData.Rmd
Modified: analysis/NewPeakPostMP.Rmd
Modified: analysis/NuclearSpecQTL.Rmd
Modified: analysis/PeakToXper.Rmd
Modified: analysis/apaQTLoverlapGWAS.Rmd
Modified: analysis/characterize_apaQTLs.Rmd
Modified: analysis/cleanupdtseq.internalpriming.Rmd
Modified: analysis/coloc_apaQTLs_protQTLs.Rmd
Modified: analysis/dif.iso.usage.leafcutter.Rmd
Modified: analysis/diff_iso_pipeline.Rmd
Modified: analysis/explainpQTLs.Rmd
Modified: analysis/explore.filters.Rmd
Modified: analysis/fixBWChromNames.Rmd
Modified: analysis/flash2mash.Rmd
Modified: analysis/initialPacBioQuant.Rmd
Modified: analysis/mispriming_approach.Rmd
Modified: analysis/overlapMolQTL.Rmd
Modified: analysis/overlapMolQTL.opposite.Rmd
Modified: analysis/overlap_qtls.Rmd
Modified: analysis/peakOverlap_oppstrand.Rmd
Modified: analysis/peakQCPPlots.Rmd
Modified: analysis/pheno.leaf.comb.Rmd
Modified: analysis/pipeline_55Ind.Rmd
Modified: analysis/swarmPlots_QTLs.Rmd
Modified: analysis/test.max2.Rmd
Modified: analysis/test.smash.Rmd
Modified: analysis/understandPeaks.Rmd
Modified: analysis/unexplainedeQTL_analysis.Rmd
Modified: code/Snakefile
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 888e7f0 | Briana Mittleman | 2019-03-20 | add examples |
| html | 7b9f73a | Briana Mittleman | 2019-03-19 | Build site. |
| Rmd | e15a531 | Briana Mittleman | 2019-03-19 | plot all tested snps |
| html | dbfcfd1 | Briana Mittleman | 2019-03-19 | Build site. |
| Rmd | b562a7e | Briana Mittleman | 2019-03-19 | add qqplots |
| html | 9aa1003 | Briana Mittleman | 2019-03-19 | Build site. |
| Rmd | d924da6 | Briana Mittleman | 2019-03-19 | add ctcf analysis |
I will look at ctcf data too see if this insulator element could act as a mechanism for apa qtls. This is in line with the kinetic model. We know CTCF binding slows polymerase. We are testing if this slow down is associated with APA as well.
The ctcf data I will use can be found at https://www.ncbi.nlm.nih.gov/pubmed/27010758
I will download the normalized phenotype file each row a binding region and each column a sample.
The regions are defined as chromosome, start, end in hg19. I will format this file so I have an ID like i do for the APA analysis.
library(tidyverse)── Attaching packages ────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.0 ✔ purrr 0.3.1
✔ tibble 2.0.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.4.0
✔ readr 1.3.1 ✔ forcats 0.4.0 Warning: package 'tibble' was built under R version 3.5.2Warning: package 'tidyr' was built under R version 3.5.2Warning: package 'purrr' was built under R version 3.5.2Warning: package 'dplyr' was built under R version 3.5.2Warning: package 'stringr' was built under R version 3.5.2Warning: package 'forcats' was built under R version 3.5.2── Conflicts ───────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(workflowr)This is workflowr version 1.2.0
Run ?workflowr for help getting startedlibrary(data.table)Warning: package 'data.table' was built under R version 3.5.2
Attaching package: 'data.table'The following objects are masked from 'package:dplyr':
between, first, lastThe following object is masked from 'package:purrr':
transposelibrary(cowplot)Warning: package 'cowplot' was built under R version 3.5.2
Attaching package: 'cowplot'The following object is masked from 'package:ggplot2':
ggsavePAS Enrichment at CTCF
First, I will look at overlap between the PAS and these CTCF sites. I can do this with deep tools by making a bed file.
CTCF2bed.py
CTCF=open("/project2/gilad/briana/threeprimeseq/data/CTCF/CTCFbinding.csv", "r")
bedFile=open("/project2/gilad/briana/threeprimeseq/data/CTCF/CTCFbindingLoc.bed", "w")
for i,ln in enumerate(CTCF):
if i >0:
chrm=ln.split(",")[0]
start=ln.split(",")[1]
end=ln.split(",")[2]
bedFile.write("%s\t%s\t%s\n"%(chrm, start, end))
bedFile.close()Deeptools plots:
TotandNucAtCTCF_DTPlot_noMPFilt.sh
#!/bin/bash
#SBATCH --job-name=TotandNucAtCTCF_DTPlot_noMPFilt
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=TotandNucAtCTCF_DTPlot_noMPFilt.out
#SBATCH --error=TotandNucAtCTCF_DTPlot_noMPFilt.err
#SBATCH --partition=bigmem2
#SBATCH --mem=100G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
computeMatrix reference-point -S /project2/gilad/briana/threeprimeseq/data/mergedBW/Total_MergedBamCoverage.bw /project2/gilad/briana/threeprimeseq/data/mergedBW/Nuclear_MergedBamCoverage.bw -R /project2/gilad/briana/threeprimeseq/data/CTCF/CTCFbindingLoc.bed -b 1000 -a 1000 -out /project2/gilad/briana/threeprimeseq/data/CTCF/TotalandNucAtCTCF.gz
plotHeatmap --sortRegions descend -m /project2/gilad/briana/threeprimeseq/data/CTCF/TotalandNucAtCTCF.gz --refPointLabel "CTCF" --plotTitle "Combined 3' at CTCF" --heatmapHeight 7 --colorMap YlGnBu -out /project2/gilad/briana/threeprimeseq/data/CTCF/TotalandNucAtCTCF.pngNo enrichemnt
Call CTCF QTL
I want to reformat the phenotypes, this is easiest in R.
CTCF=read.csv("../data/CTCF/CTCFbinding.csv", header=T) %>% mutate(ID= paste(chrm,start, end, sep=":")) %>% dplyr::select(chrm, start, end, ID, contains("NA"))
write.table(CTCF, file="../data/CTCF/CTCFbinding.pheno.bed",col.names = T, row.names = F, quote = F, sep="\t" )put on midway
#remove header
sort -k1,1 -k2,2n CTCFbinding.pheno.bed > CTCFbinding.pheno.sort.bed
#add header
bgzip CTCFbinding.pheno.sort.bed
tabix CTCFbinding.pheno.sort.bed.gz
#get the PCs
#midway1
#export PATH=/project/gilad/software/midway1/qtltools-1.0:$PATH
QTLtools pca --bed /project2/gilad/briana/threeprimeseq/data/CTCF/CTCFbinding.pheno.sort.bed.gz --scale --center --out /project2/gilad/briana/threeprimeseq/data/CTCF/CTCFbinding.pheno.sort.bed.PC.out
head -n 6 CTCFbinding.pheno.sort.bed.PC.out.pca > CTCFbinding.pheno.sort.bed.5PCs.out.pcaMake samples file:
smaplesCTCF.py
ctcf=open("/project2/gilad/briana/threeprimeseq/data/CTCF/CTCFbinding.csv", "r")
sampleFile=open("/project2/gilad/briana/threeprimeseq/data/CTCF/samples.txt", "w")
samplesVCF=open("/project2/gilad/briana/YRI_geno_hg19/vcf.samples.txt", "r")
samplesoK={}
for ln in samplesVCF:
samList=ln.split()
for i in samList:
samplesoK[i]=""
print(samplesoK)
for i, ln in enumerate(ctcf):
if i >0:
lnList=ln.split(",")
for each in lnList:
if each in samplesoK.keys():
sampleFile.write("%s\n"%(each))
else:
print("notInvcf")
sampleFile.close()
VCF file does not have these samples.
CTCFqtl_nom.sh
#!/bin/bash
#SBATCH --job-name=CTCFqtl_nom
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=CTCFqtl_nom.out
#SBATCH --error=CTCFqtl_nom.err
#SBATCH --partition=broadwl
#SBATCH --mem=12G
#SBATCH --mail-type=END
for i in $(seq 1 30)
do
/home/brimittleman/software/bin/FastQTL/bin/fastQTL.static --vcf /project2/gilad/briana/YRI_geno_hg19/allChrom.dose.filt.vcf.gz --cov /project2/gilad/briana/threeprimeseq/data/CTCF/CTCFbinding.pheno.sort.bed.5PCs.out.pca --bed /project2/gilad/briana/threeprimeseq/data/CTCF/CTCFbinding.pheno.sort.bed.gz --out /project2/gilad/briana/threeprimeseq/data/CTCF/nom/fastqtl_CTCFbinding.nominal.out --chunk $i 30 --window 5e5 --include-samples /project2/gilad/briana/threeprimeseq/data/CTCF/samples.txt
doneUse QTLs they called
Info from site:
This is the called QTLs 1% FDR threshold (q value <= 0.01) and kept only cluster variants defined as having P value within one order of magnitude to the P value of the lead variant for the same binding region.
I can make a file with the snp positions and I will look for these in my nominal APA data
ctcfQTL=read.csv("../data/CTCF/CTCFQTLS.csv")
ctcfQTL_snponly=ctcfQTL %>% dplyr::select(VARIANT_CHRM, VARIANT_POS) %>% mutate(snp_loc=paste(VARIANT_CHRM,VARIANT_POS,sep= ":")) %>% dplyr::select(snp_loc)
write.table(ctcfQTL_snponly, file="../data/CTCF/CTCFqtl_snps.txt", col.names = F, row.names = F, quote = F)Look for these snps in nominal data:
CTCFqtlinAPA.py
def main(apa, ctcfQTL, outFile):
fout=open(outFile,"w")
ctcfdic={}
for ln in open(ctcfQTL,"r"):
snp=ln.split()[0]
ctcfdic[snp]=""
for ln in open(apa, "r"):
snpApa =ln.split()[1]
if snpApa in ctcfdic.keys():
fout.write(ln)
fout.close()
if __name__ == "__main__":
import sys
fraction=sys.argv[1]
OutFile=sys.argv[2]
ctcfQTL="/project2/gilad/briana/threeprimeseq/data/CTCF/CTCFqtl_snps.txt"
if fraction=="Total":
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
else:
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
main(nomFile, ctcfQTL, OutFile) Run: run_CTCFqtlinAPA.sh
#!/bin/bash
#SBATCH --job-name=run_CTCFqtlinAPA
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=run_CTCFqtlinAPA.out
#SBATCH --error=run_CTCFqtlinAPA.err
#SBATCH --partition=broadwl
#SBATCH --mem=36G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
python CTCFqtlinAPA.py "Total" "/project2/gilad/briana/threeprimeseq/data/CTCF/CTCFQtlinTotalAPA.txt"
python CTCFqtlinAPA.py "Nuclear" "/project2/gilad/briana/threeprimeseq/data/CTCF/CTCFQtlinNuclearAPA.txt"Make empirical distribution:
I can do empirical distribution based on genes not in this set. I will make a list of the genes with] an overlap in total and in nuclear.
I can then find the matched peak numbers based on the genes that do have an overlap.
getCTCFgenes.py
apaNuc=open("/project2/gilad/briana/threeprimeseq/data/CTCF/CTCFQtlinNuclearAPA.txt", "r")
apaTot=open("/project2/gilad/briana/threeprimeseq/data/CTCF/CTCFQtlinTotalAPA.txt","r")
nucGenes=open("/project2/gilad/briana/threeprimeseq/data/CTCF/CTCFQtlinNuclearAPA_Genes.txt", "w")
totGenes=open("/project2/gilad/briana/threeprimeseq/data/CTCF/CTCFQtlinTotalAPA_Genes.txt", "w")
def overlapGenes(inFile, outFile):
#make dictionary with gene (this will have unique)
geneDic={}
for ln in inFile:
gene=ln.split()[0].split(":")[-1].split("_")[0]
if gene not in geneDic.keys():
geneDic[gene]=""
for k,v in geneDic.items():
outFile.write("%s\n"%(k))
outFile.close()
overlapGenes(apaTot, totGenes)
overlapGenes(apaNuc,nucGenes)
Plot these compared to the actuall apa QTLs.
ctcfinTot=read.table("../data/CTCF/CTCFQtlinTotalAPA.txt",stringsAsFactors = F,col.names = c("ID", "snp", "dist", "pval", "slope"))
apaTot=read.table("../data/perm_QTL_GeneLocAnno_noMP_5percov/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Total.fixed.pheno_5perc_permResBH.txt", header=T, stringsAsFactors=F)%>% drop_na()
ctcfinNuc=read.table("../data/CTCF/CTCFQtlinNuclearAPA.txt",stringsAsFactors = F,col.names = c("ID", "snp", "dist", "pval", "slope"))
apaNuc=read.table("../data/perm_QTL_GeneLocAnno_noMP_5percov/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Nuclear.fixed.pheno_5perc_permResBH.txt", header=T, stringsAsFactors=F) %>% drop_na()I need the nominal association so i am making a fair comparison
nom4apaQTLSnps.py
totQTL=open("/project2/gilad/briana/threeprimeseq/data/ApaQTLs/TotalapaQTLs.GeneLocAnno.noMP.5perc.10FDR.txt","r")
totNom=open("/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt", "r")
totNomQTL=open("/project2/gilad/briana/threeprimeseq/data/ApaQTLs/Totalapa.NomPvalAssoc4allQTL.txt", "w")
nucQTL=open("/project2/gilad/briana/threeprimeseq/data/ApaQTLs/NuclearapaQTLs.GeneLocAnno.noMP.5perc.10FDR.txt","r")
nucNom=open("/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt","r")
nucNomQTL=open("/project2/gilad/briana/threeprimeseq/data/ApaQTLs/Nuclearapa.NomPvalAssoc4allQTL.txt","w")
def allAssocAPAqtl(QTL, nom, outFile):
snpdic={}
for ln in QTL:
snp =ln.split()[5]
if snp not in snpdic.keys():
snpdic[snp]=""
for ln in nom:
snp=ln.split()[1]
if snp in snpdic.keys():
outFile.write(ln)
outFile.close()
allAssocAPAqtl(totQTL, totNom, totNomQTL)
allAssocAPAqtl(nucQTL, nucNom, nucNomQTL)
totQTLnom=read.table("../data/CTCF/Totalapa.NomPvalAssoc4allQTL.txt", stringsAsFactors = F, col.names = c("ID", "snp", "dist", "pval", "slope"))
nucQTLnom=read.table("../data/CTCF/Nuclearapa.NomPvalAssoc4allQTL.txt", stringsAsFactors = F, col.names = c("ID", "snp", "dist", "pval", "slope"))qqplot(-log10(runif(nrow(totQTLnom))),-log10(totQTLnom$pval), ylab="-log10 Tota pval", xlab="Uniform expectation", main="Total pvalue association for CTCF qtls")
points(sort(-log10(runif(nrow(ctcfinTot)))), sort(-log10(ctcfinTot$pval)),col="blue")
abline(0,1)
legend("bottomright", legend=c("ApaQTLs", "CTCF qtls"),col=c("black", "blue"), pch=16,bty = 'n')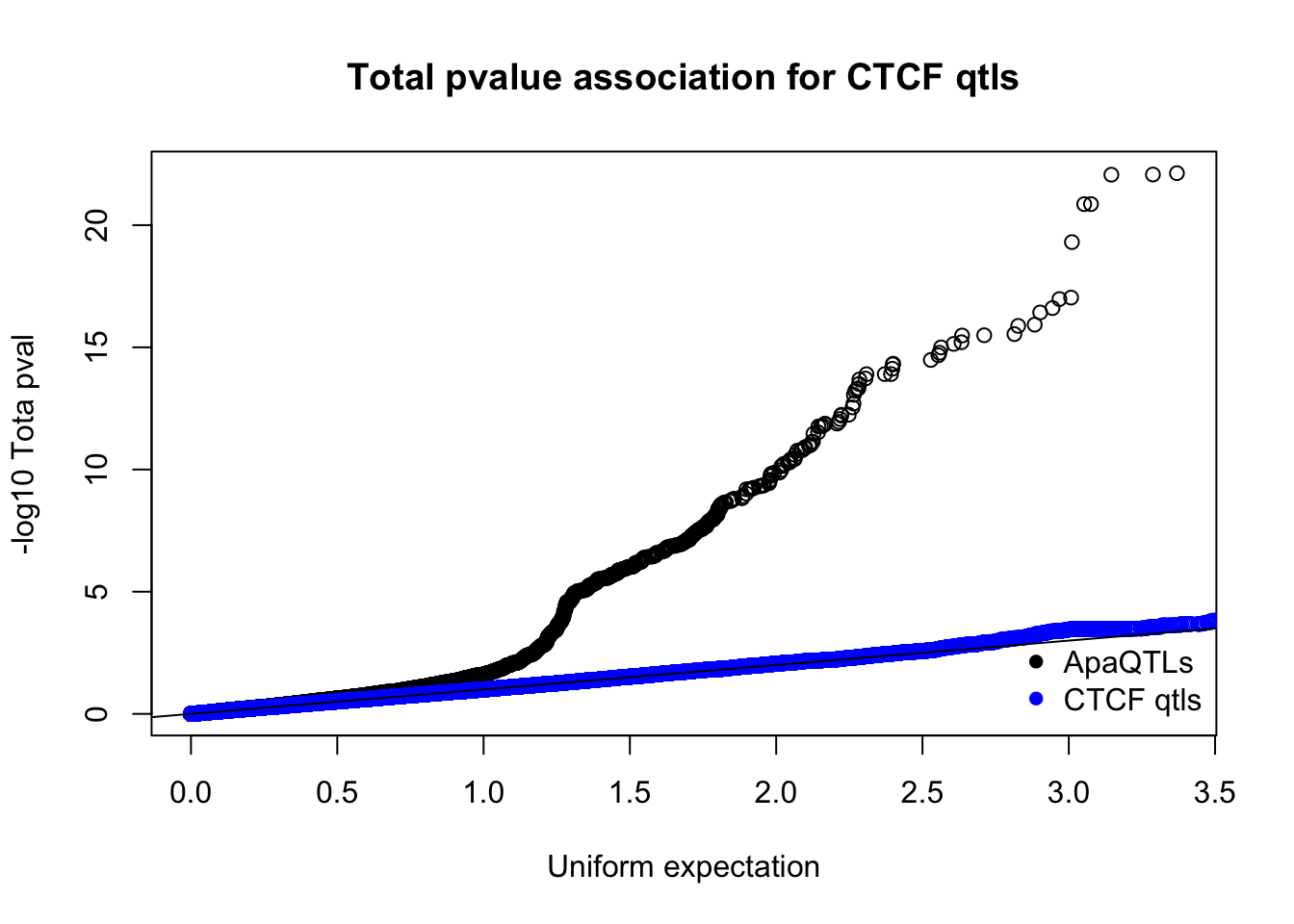
| Version | Author | Date |
|---|---|---|
| dbfcfd1 | Briana Mittleman | 2019-03-19 |
qqplot(-log10(runif(nrow(nucQTLnom))),-log10(nucQTLnom$pval), ylab="-log10 Nuclear pval", xlab="Uniform expectation", main="Nuclear pvalue association for CTCF qtls")
points(sort(-log10(runif(nrow(ctcfinNuc)))), sort(-log10(ctcfinNuc$pval)),col="blue")
abline(0,1)
legend("bottomright", legend=c("ApaQTLs", "CTCF qtls"),col=c("black", "blue"), pch=16,bty = 'n')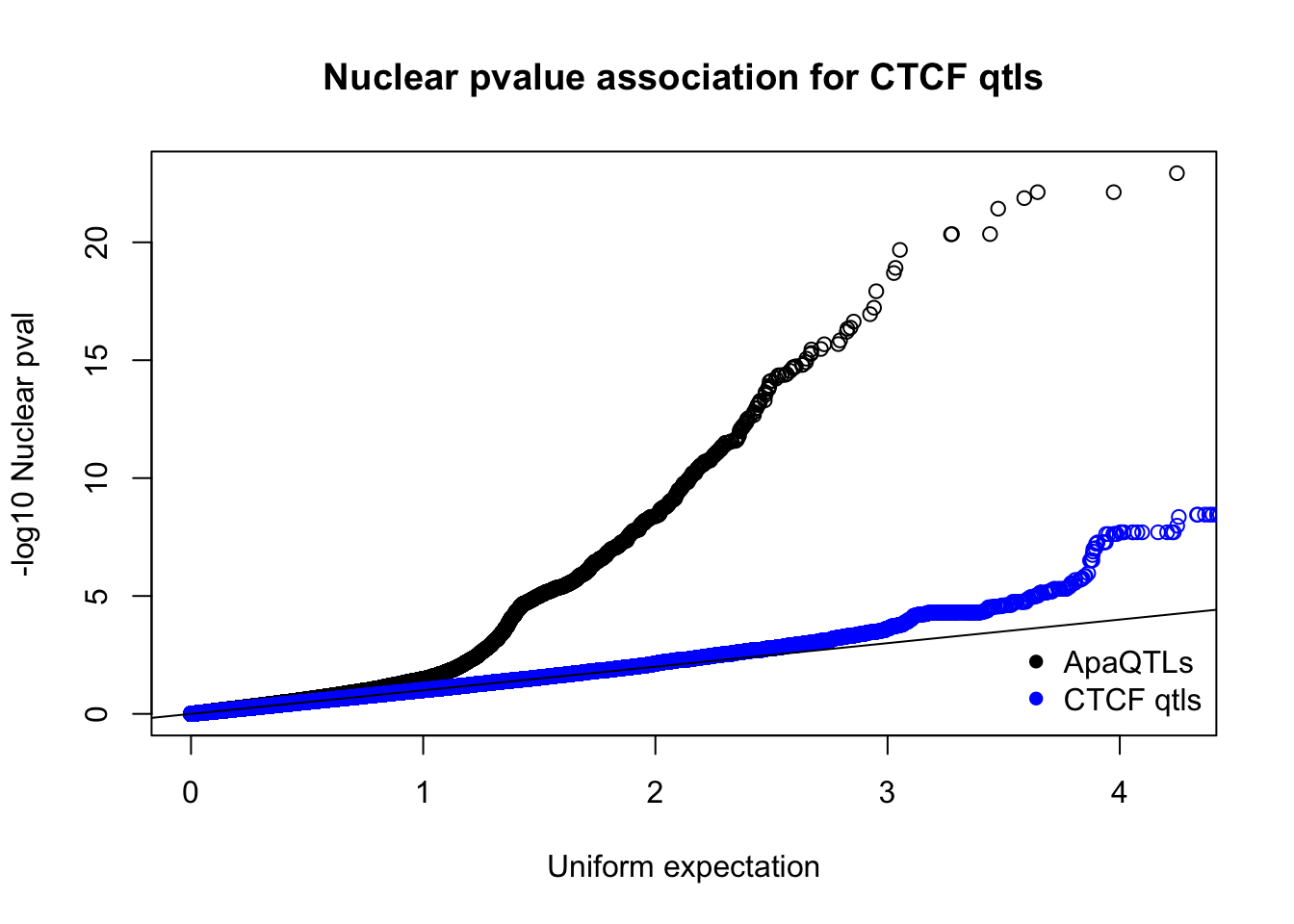
| Version | Author | Date |
|---|---|---|
| dbfcfd1 | Briana Mittleman | 2019-03-19 |
Use different black line, dont condition on apaQTLs look at every tested snp. I can sort the pvalues in the nominal file then sample every 100th value to deal wilth how large the vector is:
Python interactively
totNom="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
nucNom="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
import numpy as np
import pandas as pd
Nomnames=["ID", "snp", "dist", "pval", "slope"]
nomDF=pd.read_table(totNom, sep=" ", names=Nomnames, header=None)
a=nomDF.iloc[:,3]
sorted=np.sort(a)
final=sorted[0::100]
np.savetxt('/project2/gilad/briana/threeprimeseq/data/CTCF/TotNomPvalsOneHund.out', final)
nomNucDF=pd.read_table(nucNom, sep=" ", names=Nomnames, header=None)
b=nomNucDF.iloc[:,3]
sortedNuc=np.sort(b)
finalNuc=sortedNuc[0::100]
np.savetxt('/project2/gilad/briana/threeprimeseq/data/CTCF/NucNomPvalsOneHund.out', finalNuc) use this:
totnomPvalsmall=read.table("../data/CTCF/TotNomPvalsOneHund.out", header = F, stringsAsFactors = F, col.names = c("pval"))
qqplot(-log10(runif(nrow(totnomPvalsmall))),-log10(totnomPvalsmall$pval), ylab="-log10 Total pval", xlab="Uniform expectation", main="Total pvalue association for CTCF qtls")
points(sort(-log10(runif(nrow(ctcfinTot)))), sort(-log10(ctcfinTot$pval)),col="blue")
abline(0,1)
legend("bottomright", legend=c("1/100 Apa SNPs", "CTCF qtls"),col=c("black", "blue"), pch=16,bty = 'n')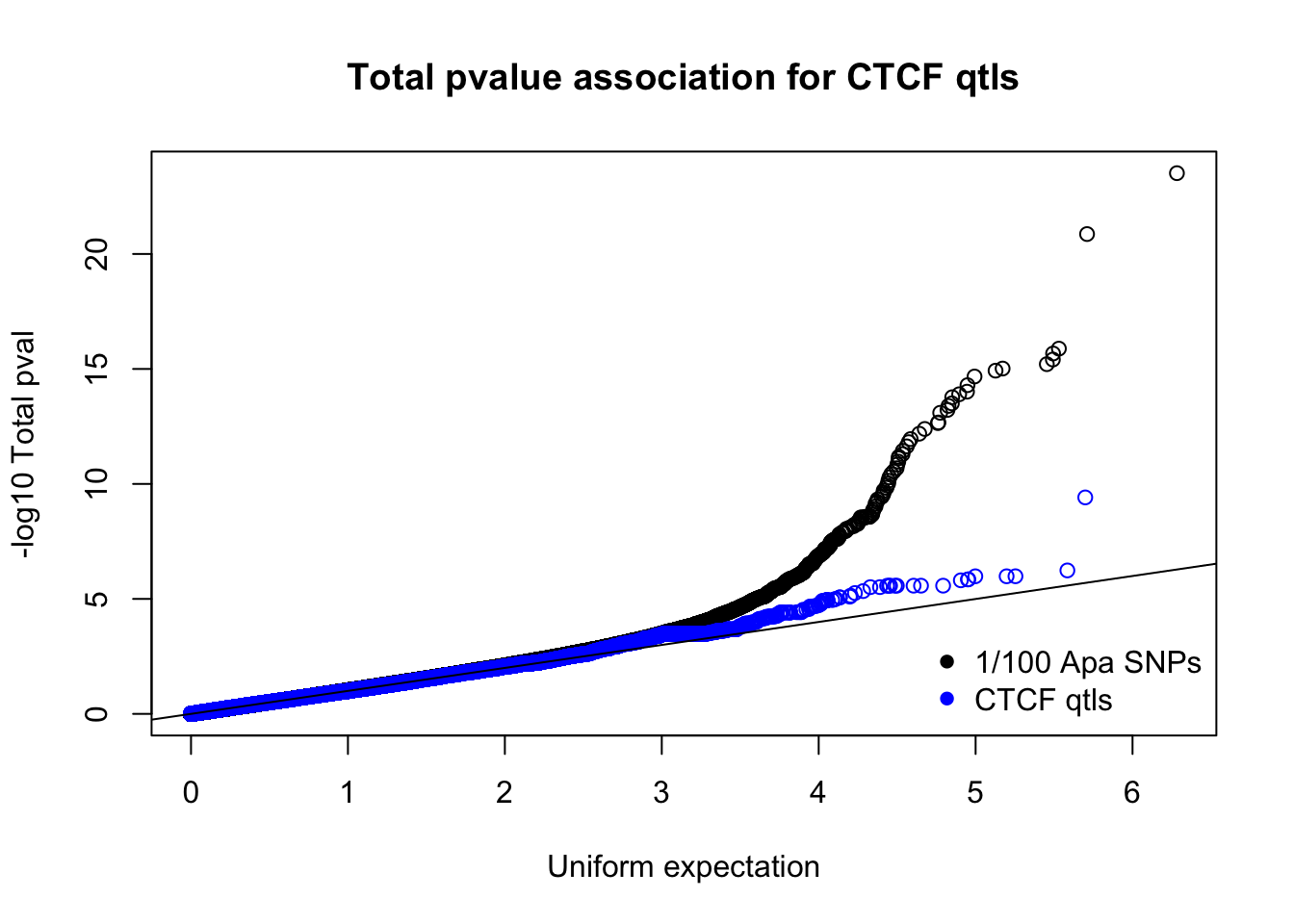
| Version | Author | Date |
|---|---|---|
| 7b9f73a | Briana Mittleman | 2019-03-19 |
nucnomPvalsmall=read.table("../data/CTCF/NucNomPvalsOneHund.out", header = F, stringsAsFactors = F, col.names = c("pval"))
qqplot(-log10(runif(nrow(nucnomPvalsmall))),-log10(nucnomPvalsmall$pval), ylab="-log10 Nuclear pval", xlab="Uniform expectation", main="Nuclear pvalue association for CTCF qtls")
points(sort(-log10(runif(nrow(ctcfinNuc)))), sort(-log10(ctcfinNuc$pval)),col="blue")
abline(0,1)
legend("bottomright", legend=c("1/100 Apa SNPs", "CTCF qtls"),col=c("black", "blue"), pch=16,bty = 'n')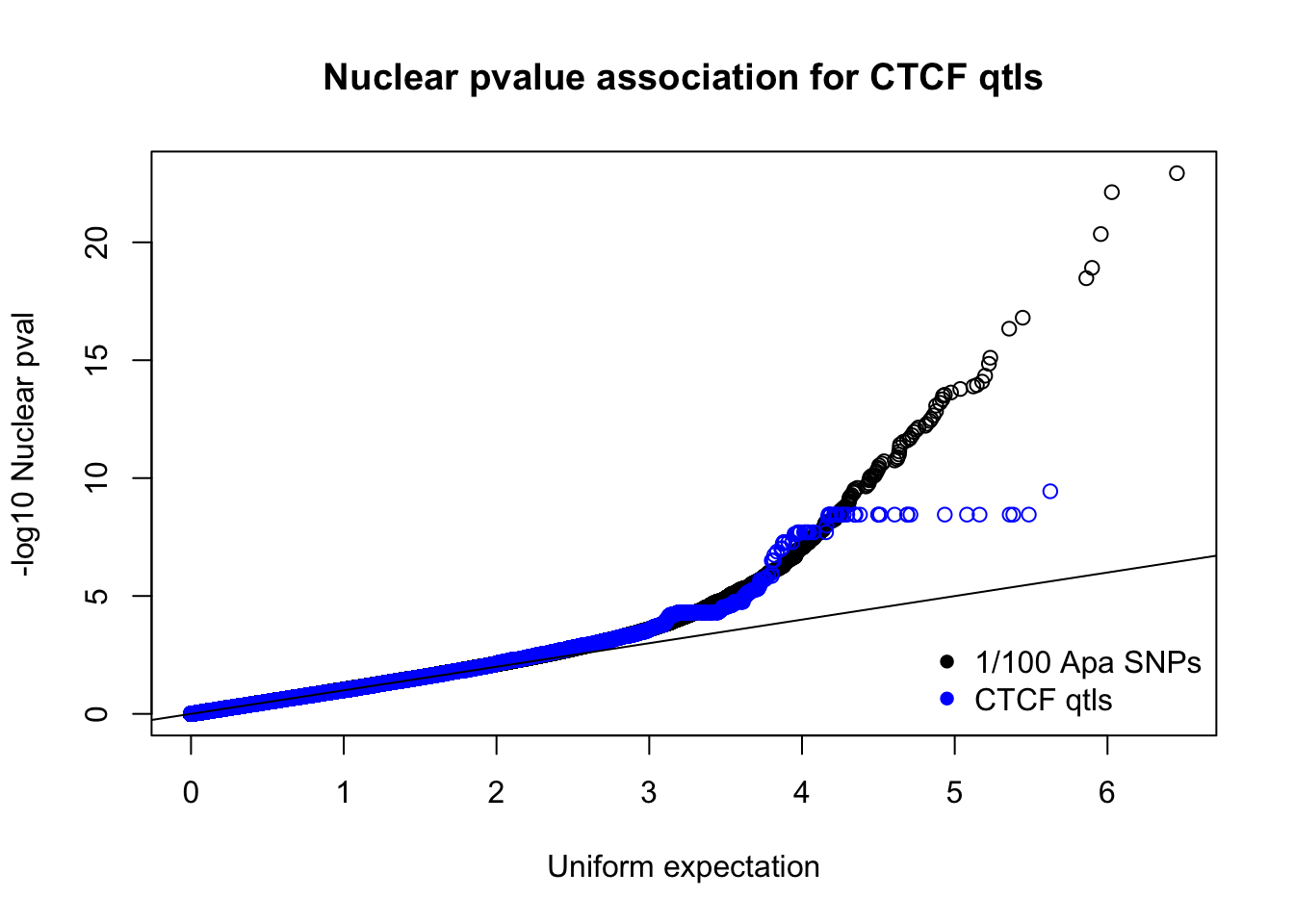
| Version | Author | Date |
|---|---|---|
| 7b9f73a | Briana Mittleman | 2019-03-19 |
Look at some examples
Look at the points in the nuclear fraction that are -log10(pval)>5.
ctcfinNuc %>% filter(-log10(pval)>5) %>% arrange(pval) ID snp dist
1 1:146735002:146735080:CHD1L_-_peak9190 1:146699958 -35045
2 14:60611650:60611732:DHRS7_+_peak48511 14:60641082 29431
3 14:60611650:60611732:DHRS7_+_peak48511 14:60612714 1063
4 14:60611650:60611732:DHRS7_+_peak48511 14:60612766 1115
5 14:60611650:60611732:DHRS7_+_peak48511 14:60612963 1312
6 14:60611650:60611732:DHRS7_+_peak48511 14:60613891 2240
7 14:60611650:60611732:DHRS7_+_peak48511 14:60614011 2360
8 14:60611650:60611732:DHRS7_+_peak48511 14:60614176 2525
9 14:60611650:60611732:DHRS7_+_peak48511 14:60615215 3564
10 14:60611650:60611732:DHRS7_+_peak48511 14:60615616 3965
11 14:60611650:60611732:DHRS7_+_peak48511 14:60615625 3974
12 14:60611650:60611732:DHRS7_+_peak48511 14:60615722 4071
13 14:60611650:60611732:DHRS7_+_peak48511 14:60615914 4263
14 14:60611650:60611732:DHRS7_+_peak48511 14:60617198 5547
15 14:60611650:60611732:DHRS7_+_peak48511 14:60617238 5587
16 14:60611650:60611732:DHRS7_+_peak48511 14:60617333 5682
17 14:60611650:60611732:DHRS7_+_peak48511 14:60617639 5988
18 14:60611650:60611732:DHRS7_+_peak48511 14:60617640 5989
19 14:60611650:60611732:DHRS7_+_peak48511 14:60618745 7094
20 14:60611650:60611732:DHRS7_+_peak48511 14:60618812 7161
21 14:60611650:60611732:DHRS7_+_peak48511 14:60619111 7460
22 14:60611650:60611732:DHRS7_+_peak48511 14:60620050 8399
23 14:60611650:60611732:DHRS7_+_peak48511 14:60620259 8608
24 14:60611650:60611732:DHRS7_+_peak48511 14:60622082 10431
25 14:60611650:60611732:DHRS7_+_peak48511 14:60625039 13388
26 14:60611650:60611732:DHRS7_+_peak48511 14:60628608 16957
27 14:60611650:60611732:DHRS7_+_peak48511 14:60634111 22460
28 14:60611650:60611732:DHRS7_+_peak48511 14:60638815 27164
29 14:60611650:60611732:DHRS7_+_peak48511 14:60638926 27275
30 14:60611650:60611732:DHRS7_+_peak48511 14:60619317 7666
31 14:60611650:60611732:DHRS7_+_peak48511 14:60617696 6045
32 22:39800489:39800565:TAB1_-_peak104641 22:39822116 21626
33 14:60611650:60611732:DHRS7_+_peak48511 14:60607059 -4592
34 14:60611650:60611732:DHRS7_+_peak48511 14:60607765 -3886
35 14:60611650:60611732:DHRS7_+_peak48511 14:60607985 -3666
36 14:60611650:60611732:DHRS7_+_peak48511 14:60608457 -3194
37 14:60611650:60611732:DHRS7_+_peak48511 14:60613564 1913
38 14:60611650:60611732:DHRS7_+_peak48511 14:60614038 2387
39 14:60611650:60611732:DHRS7_+_peak48511 14:60616042 4391
40 14:60611650:60611732:DHRS7_+_peak48511 14:60619179 7528
41 14:60611650:60611732:DHRS7_+_peak48511 14:60619356 7705
42 14:60611650:60611732:DHRS7_+_peak48511 14:60623676 12025
43 14:60611650:60611732:DHRS7_+_peak48511 14:60623741 12090
44 14:60611650:60611732:DHRS7_+_peak48511 14:60634585 22934
45 1:15981871:15981942:DDI2_-_peak1257 1:15920821 -61051
46 22:39800489:39800565:TAB1_-_peak104641 22:39793766 -6724
47 22:39800489:39800565:TAB1_-_peak104641 22:39794124 -6366
48 22:39800489:39800565:TAB1_-_peak104641 22:39793079 -7411
49 22:39800489:39800565:TAB1_-_peak104641 22:39797987 -2503
50 22:39800489:39800565:TAB1_-_peak104641 22:39798429 -2061
51 1:93672606:93672687:CCDC18_-_peak6813 1:93752982 80375
52 1:93672606:93672687:CCDC18_-_peak6813 1:93753932 81325
53 1:93672606:93672687:CCDC18_-_peak6813 1:93756886 84279
54 1:93672606:93672687:CCDC18_-_peak6813 1:93750571 77964
55 1:93672606:93672687:CCDC18_-_peak6813 1:93749072 76465
56 1:93672606:93672687:CCDC18_-_peak6813 1:93720597 47990
57 22:39800489:39800565:TAB1_-_peak104641 22:39823015 22525
58 1:15980902:15980984:DDI2_-_peak1256 1:15923726 -57177
59 22:39800489:39800565:TAB1_-_peak104641 22:39810379 9889
60 1:15981871:15981942:DDI2_-_peak1257 1:15923726 -58146
61 5:177037192:177037330:B4GALT7_-_peak135486 5:176798306 -238887
62 22:39800489:39800565:TAB1_-_peak104641 22:39829736 29246
63 22:39800489:39800565:TAB1_-_peak104641 22:39826788 26298
64 22:39800489:39800565:TAB1_-_peak104641 22:39827553 27063
65 22:39800489:39800565:TAB1_-_peak104641 22:39794241 -6249
66 1:146767359:146767443:CHD1L_-_peak9202 1:146699958 -67402
67 22:39800489:39800565:TAB1_-_peak104641 22:39797178 -3312
68 22:39800489:39800565:TAB1_-_peak104641 22:39784845 -15645
69 20:30753271:30753353:TM9SF4_-_peak98455 20:31037635 284363
70 20:30753271:30753353:TM9SF4_-_peak98455 20:31028127 274855
71 16:2710351:2710433:ERVK13-1_+_peak59006 16:2710798 446
72 20:30753271:30753353:TM9SF4_-_peak98455 20:31002352 249080
73 20:30753271:30753353:TM9SF4_-_peak98455 20:30995118 241846
74 20:30753271:30753353:TM9SF4_-_peak98455 20:31012361 259089
75 20:30753271:30753353:TM9SF4_-_peak98455 20:31029615 276343
76 6:18249913:18250027:DEK_+_peak137339 6:18707766 457852
77 6:18249913:18250027:DEK_+_peak137339 6:18707584 457670
78 6:18249913:18250027:DEK_+_peak137339 6:18704706 454792
79 6:18249913:18250027:DEK_+_peak137339 6:18702517 452603
80 6:18249913:18250027:DEK_+_peak137339 6:18702152 452238
81 6:18249913:18250027:DEK_+_peak137339 6:18701468 451554
82 1:93672606:93672687:CCDC18_-_peak6813 1:93770372 97765
83 1:93672606:93672687:CCDC18_-_peak6813 1:93790645 118038
84 1:93672606:93672687:CCDC18_-_peak6813 1:93780618 108011
85 1:93672606:93672687:CCDC18_-_peak6813 1:93798391 125784
86 1:93672606:93672687:CCDC18_-_peak6813 1:93799644 127037
87 1:93672606:93672687:CCDC18_-_peak6813 1:93807262 134655
88 1:93672606:93672687:CCDC18_-_peak6813 1:93809710 137103
89 1:93672606:93672687:CCDC18_-_peak6813 1:93814825 142218
90 20:30753271:30753353:TM9SF4_-_peak98455 20:31042169 288897
91 20:30753271:30753353:TM9SF4_-_peak98455 20:31033574 280302
92 20:30753271:30753353:TM9SF4_-_peak98455 20:31036873 283601
93 20:30753271:30753353:TM9SF4_-_peak98455 20:31039688 286416
94 16:2710351:2710433:ERVK13-1_+_peak59006 16:2711014 662
95 16:2710351:2710433:ERVK13-1_+_peak59006 16:2710153 -199
96 16:2710351:2710433:ERVK13-1_+_peak59006 16:2711316 964
97 16:2710351:2710433:ERVK13-1_+_peak59006 16:2711368 1016
98 1:93803671:93803804:CCDC18-AS1_+_peak6890 1:93809710 6038
99 1:93803671:93803804:CCDC18-AS1_+_peak6890 1:93814825 11153
100 1:93803671:93803804:CCDC18-AS1_+_peak6890 1:93807262 3590
101 1:93803671:93803804:CCDC18-AS1_+_peak6890 1:93799644 -4028
102 1:93803671:93803804:CCDC18-AS1_+_peak6890 1:93798391 -5281
103 1:93803671:93803804:CCDC18-AS1_+_peak6890 1:93790645 -13027
104 5:176799516:176799598:RGS14_-_peak135435 5:176798306 -1211
105 1:93803671:93803804:CCDC18-AS1_+_peak6890 1:93780618 -23054
106 1:93803671:93803804:CCDC18-AS1_+_peak6890 1:93770372 -33300
107 16:2710351:2710433:ERVK13-1_+_peak59006 16:2714986 4634
108 16:2710351:2710433:ERVK13-1_+_peak59006 16:2717023 6671
109 16:2710351:2710433:ERVK13-1_+_peak59006 16:2717497 7145
110 13:74326901:74326981:KLF12_+_peak44717 13:74318612 -8290
111 13:74326901:74326981:KLF12_+_peak44717 13:74318497 -8405
pval slope
1 3.62229e-10 1.393670
2 3.52920e-09 0.692925
3 3.53939e-09 0.692823
4 3.53939e-09 0.692823
5 3.53939e-09 0.692823
6 3.53939e-09 0.692823
7 3.53939e-09 0.692823
8 3.53939e-09 0.692823
9 3.53939e-09 0.692823
10 3.53939e-09 0.692823
11 3.53939e-09 0.692823
12 3.53939e-09 0.692823
13 3.53939e-09 0.692823
14 3.53939e-09 0.692823
15 3.53939e-09 0.692823
16 3.53939e-09 0.692823
17 3.53939e-09 0.692823
18 3.53939e-09 0.692823
19 3.53939e-09 0.692823
20 3.53939e-09 0.692823
21 3.53939e-09 0.692823
22 3.53939e-09 0.692823
23 3.53939e-09 0.692823
24 3.53939e-09 0.692823
25 3.53939e-09 0.692823
26 3.53939e-09 0.692823
27 3.53939e-09 0.692823
28 3.53939e-09 0.692823
29 3.53939e-09 0.692823
30 3.55694e-09 0.692890
31 4.45865e-09 0.699921
32 1.04389e-08 1.079260
33 1.98846e-08 0.677083
34 1.98846e-08 0.677083
35 1.98846e-08 0.677083
36 1.98846e-08 0.677083
37 1.98846e-08 0.677083
38 1.98846e-08 0.677083
39 1.98846e-08 0.677083
40 1.98846e-08 0.677083
41 1.98846e-08 0.677083
42 1.98846e-08 0.677083
43 1.98846e-08 0.677083
44 1.98846e-08 0.677083
45 2.35186e-08 1.061530
46 2.35643e-08 1.073150
47 2.35643e-08 1.073150
48 2.36045e-08 1.073150
49 2.36341e-08 1.072740
50 2.36341e-08 1.072740
51 5.24348e-08 0.871191
52 5.24348e-08 0.871191
53 5.24410e-08 0.871272
54 5.25659e-08 0.871105
55 5.26536e-08 0.870951
56 5.30592e-08 0.869408
57 5.93198e-08 1.077070
58 6.09364e-08 -0.786164
59 9.32737e-08 1.121110
60 1.01204e-07 1.132390
61 1.31721e-07 -1.522240
62 1.82782e-07 1.011320
63 3.08979e-07 1.041260
64 3.08979e-07 1.041260
65 3.15015e-07 1.025880
66 1.08955e-06 -1.037160
67 1.42563e-06 1.068450
68 1.43352e-06 1.068570
69 1.77267e-06 -1.286020
70 1.86739e-06 -1.274550
71 1.93581e-06 -0.630680
72 1.93724e-06 -1.283520
73 2.01512e-06 -1.275480
74 2.06938e-06 -1.273500
75 2.09949e-06 -1.260120
76 2.79112e-06 1.170140
77 2.82922e-06 1.169430
78 3.33811e-06 1.159070
79 3.95584e-06 1.147640
80 4.24786e-06 1.142700
81 4.37666e-06 1.140620
82 4.93437e-06 0.695754
83 4.93964e-06 0.694792
84 4.93966e-06 0.695253
85 4.95487e-06 0.694673
86 4.96110e-06 0.694656
87 4.97752e-06 0.694586
88 4.98380e-06 0.694569
89 4.98624e-06 0.694585
90 5.42251e-06 -1.168040
91 5.43783e-06 -1.166330
92 5.49858e-06 -1.166060
93 5.49858e-06 -1.166060
94 6.39555e-06 -0.608443
95 6.41954e-06 -0.608339
96 6.42862e-06 -0.608311
97 6.42862e-06 -0.608311
98 7.13540e-06 0.668228
99 7.13642e-06 0.668253
100 7.13879e-06 0.668202
101 7.15348e-06 0.668135
102 7.15691e-06 0.668109
103 7.18001e-06 0.668065
104 7.19651e-06 -1.343850
105 7.28212e-06 0.668147
106 7.39875e-06 0.668195
107 8.83098e-06 -0.603066
108 8.90845e-06 -0.603693
109 8.99811e-06 -0.604063
110 9.91263e-06 1.064890
111 9.97338e-06 1.061920Top genes with effects:
CHD1L
DHRS7
DDI2
TAB1
CCDC18 (CCDC18-AS1)
B4GALt7
TMSF4
ERVK13-1
DEK
RGS14
KLF12
I will see if any of these have a called apaQTL
qtlNames=c("ID")
apaQTLsNuc=read.table("../data/ApaQTLs/NuclearapaQTLs.GeneLocAnno.noMP.5perc.10FDR.txt", stringsAsFactors = F, col.names = c("pid", "nvar", "shape1", "shape2", "dummy", "sid", "dist", "npval", "slope", "ppval", "bpval","bh"))
apaQTLsTot=read.table("../data/ApaQTLs/TotalapaQTLs.GeneLocAnno.noMP.5perc.10FDR.txt", stringsAsFactors = F, col.names = c("pid", "nvar", "shape1", "shape2", "dummy", "sid", "dist", "npval", "slope", "ppval", "bpval","bh"))CHD1l:
1:146735002:146735080:CHD1L_-_peak9190- 1:146728013
1:146767359:146767443:CHD1L_-_peak9202 1:146748962
DHRS7:
14:60611650:60611732:DHRS7_+_peak48511
I should add the ctcfQTLs as a bedfile:
ctcfQTL2bed.py
inQTL=open("/project2/gilad/briana/threeprimeseq/data/CTCF/CTCFQTLS.csv"."r")
outBed=open("/project2/gilad/briana/threeprimeseq/data/CTCF/CTCFQTL.bed","w")
for i, ln in enumerate(inQTL):
if i >0:
chrom=ln.split(",")[0]
start=ln.split(",")[1]
end=ln.split(",")[2]
id=chrom + ":" + start + ":" + end
varEnd=int(ln.split(",")[4])
varStart=varEnd-1
varChrom=ln.split(",")[3]
outBed.write("%s\t%d\t%d\t%s\n"%(varChrom, varStart, varEnd, id))
outBed.close() Intersect this with out QTLs and all of those in LD with ours:
interactively
allOverlapQTL='/project2/gilad/briana/threeprimeseq/data/GWAS_overlap_processed/AllOverlapSnps.bed'
import pybedtools as pybedtools
ctcf_file=open("/project2/gilad/briana/threeprimeseq/data/CTCF/CTCFQTL.bed","r")
ctcf=pybedtools.BedTool(ctcf_file)
snps_file=open(allOverlapQTL, "r")
snps=pybedtools.BedTool(snps_file)
snpOverctcf=snps.intersect(ctcf, wa=True,wb=True)
snpOverctcf.saveas("/project2/gilad/briana/threeprimeseq/data/CTCF/CTCFqtlOverlapAPAqtls.txt")
One interesting exaple
ERVK13-1 chr16:2,709,806-2,712,090
CTCF site between 2 peaks. overlap with CTCF qtl and nuclear qtl associated with downstream QTL
16 2710797 2710798
the qtl is snp_16_2710771
TAB1
chr22:39,792,017-39,826,801 22,39795500,39795832,22,39822116,rs5757667,A,G,AA=G;AN=102;AC=73;VT=SNP,0.871868708751,3.54445306103e-09,-1.2411307501,0.715823195148,0.121500443991,2.055844e-06
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.14.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] cowplot_0.9.4 data.table_1.12.0 workflowr_1.2.0
[4] forcats_0.4.0 stringr_1.4.0 dplyr_0.8.0.1
[7] purrr_0.3.1 readr_1.3.1 tidyr_0.8.3
[10] tibble_2.0.1 ggplot2_3.1.0 tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] Rcpp_1.0.0 cellranger_1.1.0 plyr_1.8.4 pillar_1.3.1
[5] compiler_3.5.1 git2r_0.24.0 tools_3.5.1 digest_0.6.18
[9] lubridate_1.7.4 jsonlite_1.6 evaluate_0.13 nlme_3.1-137
[13] gtable_0.2.0 lattice_0.20-38 pkgconfig_2.0.2 rlang_0.3.1
[17] cli_1.0.1 rstudioapi_0.9.0 yaml_2.2.0 haven_2.1.0
[21] xfun_0.5 withr_2.1.2 xml2_1.2.0 httr_1.4.0
[25] knitr_1.21 hms_0.4.2 generics_0.0.2 fs_1.2.6
[29] rprojroot_1.3-2 grid_3.5.1 tidyselect_0.2.5 glue_1.3.0
[33] R6_2.4.0 readxl_1.3.0 rmarkdown_1.11 modelr_0.1.4
[37] magrittr_1.5 whisker_0.3-2 backports_1.1.3 scales_1.0.0
[41] htmltools_0.3.6 rvest_0.3.2 assertthat_0.2.0 colorspace_1.4-0
[45] stringi_1.3.1 lazyeval_0.2.1 munsell_0.5.0 broom_0.5.1
[49] crayon_1.3.4