Empirical Distribution for overlap
Briana Mittleman
3/3/2019
Last updated: 2019-03-19
Checks: 6 0
Knit directory: threeprimeseq/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.2.0). The Report tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(12345) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/.DS_Store
Ignored: data/perm_QTL_trans_noMP_5percov/
Ignored: output/.DS_Store
Untracked files:
Untracked: KalistoAbundance18486.txt
Untracked: analysis/4suDataIGV.Rmd
Untracked: analysis/DirectionapaQTL.Rmd
Untracked: analysis/EvaleQTLs.Rmd
Untracked: analysis/YL_QTL_test.Rmd
Untracked: analysis/groSeqAnalysis.Rmd
Untracked: analysis/ncbiRefSeq_sm.sort.mRNA.bed
Untracked: analysis/snake.config.notes.Rmd
Untracked: analysis/verifyBAM.Rmd
Untracked: analysis/verifybam_dubs.Rmd
Untracked: code/PeaksToCoverPerReads.py
Untracked: code/strober_pc_pve_heatmap_func.R
Untracked: data/18486.genecov.txt
Untracked: data/APApeaksYL.total.inbrain.bed
Untracked: data/AllPeak_counts/
Untracked: data/ApaQTLs/
Untracked: data/ApaQTLs_otherPhen/
Untracked: data/ChromHmmOverlap/
Untracked: data/DistTXN2Peak_genelocAnno/
Untracked: data/EmpiricalDists/
Untracked: data/FeatureoverlapPeaks/
Untracked: data/GM12878.chromHMM.bed
Untracked: data/GM12878.chromHMM.txt
Untracked: data/GWAS_overlap/
Untracked: data/LianoglouLCL/
Untracked: data/LocusZoom/
Untracked: data/LocusZoom_Unexp/
Untracked: data/LocusZoom_proc/
Untracked: data/MatchedSnps/
Untracked: data/NucSpecQTL/
Untracked: data/NuclearApaQTLs.txt
Untracked: data/PeakCounts/
Untracked: data/PeakCounts_noMP_5perc/
Untracked: data/PeakCounts_noMP_genelocanno/
Untracked: data/PeakUsage/
Untracked: data/PeakUsage_noMP/
Untracked: data/PeakUsage_noMP_GeneLocAnno/
Untracked: data/PeaksUsed/
Untracked: data/PeaksUsed_noMP_5percCov/
Untracked: data/PolyA_DB/
Untracked: data/QTL_overlap/
Untracked: data/RNAdecay/
Untracked: data/RNAkalisto/
Untracked: data/RefSeq_annotations/
Untracked: data/Replicates_usage/
Untracked: data/Signal_Loc/
Untracked: data/TotalApaQTLs.txt
Untracked: data/Totalpeaks_filtered_clean.bed
Untracked: data/UnderstandPeaksQC/
Untracked: data/WASP_STAT/
Untracked: data/YL-SP-18486-T-combined-genecov.txt
Untracked: data/YL-SP-18486-T_S9_R1_001-genecov.txt
Untracked: data/YL_QTL_test/
Untracked: data/apaExamp/
Untracked: data/apaExamp_proc/
Untracked: data/apaQTL_examp_noMP/
Untracked: data/bedgraph_peaks/
Untracked: data/bin200.5.T.nuccov.bed
Untracked: data/bin200.Anuccov.bed
Untracked: data/bin200.nuccov.bed
Untracked: data/clean_peaks/
Untracked: data/comb_map_stats.csv
Untracked: data/comb_map_stats.xlsx
Untracked: data/comb_map_stats_39ind.csv
Untracked: data/combined_reads_mapped_three_prime_seq.csv
Untracked: data/diff_iso_GeneLocAnno/
Untracked: data/diff_iso_proc/
Untracked: data/diff_iso_trans/
Untracked: data/eQTL_inAPA/
Untracked: data/eQTLs_Lietal/
Untracked: data/ensemble_to_genename.txt
Untracked: data/example_gene_peakQuant/
Untracked: data/explainProtVar/
Untracked: data/filtPeakOppstrand_cov_noMP_GeneLocAnno_5perc/
Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.bed
Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.noties.bed
Untracked: data/first50lines_closest.txt
Untracked: data/gencov.test.csv
Untracked: data/gencov.test.txt
Untracked: data/gencov_zero.test.csv
Untracked: data/gencov_zero.test.txt
Untracked: data/gene_cov/
Untracked: data/joined
Untracked: data/leafcutter/
Untracked: data/merged_combined_YL-SP-threeprimeseq.bg
Untracked: data/molPheno_noMP/
Untracked: data/mol_overlap/
Untracked: data/mol_pheno/
Untracked: data/nom_QTL/
Untracked: data/nom_QTL_opp/
Untracked: data/nom_QTL_trans/
Untracked: data/nuc6up/
Untracked: data/nuc_10up/
Untracked: data/other_qtls/
Untracked: data/pQTL_inAPA/
Untracked: data/pQTL_otherphen/
Untracked: data/pacbio_cov/
Untracked: data/peakPerRefSeqGene/
Untracked: data/peaks4DT/
Untracked: data/perm_QTL/
Untracked: data/perm_QTL_GeneLocAnno_noMP_5percov/
Untracked: data/perm_QTL_GeneLocAnno_noMP_5percov_3UTR/
Untracked: data/perm_QTL_diffWindow/
Untracked: data/perm_QTL_opp/
Untracked: data/perm_QTL_trans/
Untracked: data/perm_QTL_trans_filt/
Untracked: data/protAndAPAAndExplmRes.Rda
Untracked: data/protAndAPAlmRes.Rda
Untracked: data/protAndExpressionlmRes.Rda
Untracked: data/reads_mapped_three_prime_seq.csv
Untracked: data/smash.cov.results.bed
Untracked: data/smash.cov.results.csv
Untracked: data/smash.cov.results.txt
Untracked: data/smash_testregion/
Untracked: data/ssFC200.cov.bed
Untracked: data/temp.file1
Untracked: data/temp.file2
Untracked: data/temp.gencov.test.txt
Untracked: data/temp.gencov_zero.test.txt
Untracked: data/threePrimeSeqMetaData.csv
Untracked: data/threePrimeSeqMetaData55Ind.txt
Untracked: data/threePrimeSeqMetaData55Ind.xlsx
Untracked: data/threePrimeSeqMetaData55Ind_noDup.txt
Untracked: data/threePrimeSeqMetaData55Ind_noDup.xlsx
Untracked: data/threePrimeSeqMetaData55Ind_noDup_WASPMAP.txt
Untracked: data/threePrimeSeqMetaData55Ind_noDup_WASPMAP.xlsx
Untracked: manuscript/
Untracked: output/LZ/
Untracked: output/deeptools_plots/
Untracked: output/picard/
Untracked: output/plots/
Untracked: output/qual.fig2.pdf
Unstaged changes:
Modified: analysis/28ind.peak.explore.Rmd
Modified: analysis/CompareLianoglouData.Rmd
Modified: analysis/NewPeakPostMP.Rmd
Modified: analysis/NuclearSpecQTL.Rmd
Modified: analysis/PeakToXper.Rmd
Modified: analysis/apaQTLoverlapGWAS.Rmd
Modified: analysis/characterize_apaQTLs.Rmd
Modified: analysis/cleanupdtseq.internalpriming.Rmd
Modified: analysis/coloc_apaQTLs_protQTLs.Rmd
Modified: analysis/dif.iso.usage.leafcutter.Rmd
Modified: analysis/diff_iso_pipeline.Rmd
Modified: analysis/explainpQTLs.Rmd
Modified: analysis/explore.filters.Rmd
Modified: analysis/fixBWChromNames.Rmd
Modified: analysis/flash2mash.Rmd
Modified: analysis/initialPacBioQuant.Rmd
Modified: analysis/mispriming_approach.Rmd
Modified: analysis/overlapMolQTL.Rmd
Modified: analysis/overlapMolQTL.opposite.Rmd
Modified: analysis/overlap_qtls.Rmd
Modified: analysis/peakOverlap_oppstrand.Rmd
Modified: analysis/peakQCPPlots.Rmd
Modified: analysis/pheno.leaf.comb.Rmd
Modified: analysis/pipeline_55Ind.Rmd
Modified: analysis/swarmPlots_QTLs.Rmd
Modified: analysis/test.max2.Rmd
Modified: analysis/test.smash.Rmd
Modified: analysis/understandPeaks.Rmd
Modified: analysis/unexplainedeQTL_analysis.Rmd
Modified: code/Snakefile
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | e83b47b | Briana Mittleman | 2019-03-19 | add total vs nuclear p not e |
| html | 57f22b1 | Briana Mittleman | 2019-03-13 | Build site. |
| Rmd | 068ae89 | Briana Mittleman | 2019-03-13 | add pqtl analysis |
| html | f696384 | Briana Mittleman | 2019-03-13 | Build site. |
| Rmd | 01c22a6 | Briana Mittleman | 2019-03-13 | eQTL and unexp eQTL res |
| html | aa20236 | Briana Mittleman | 2019-03-13 | Build site. |
| Rmd | e556f63 | Briana Mittleman | 2019-03-13 | eQTL and unexp eQTL res |
library(workflowr)This is workflowr version 1.2.0
Run ?workflowr for help getting startedlibrary(tidyverse)── Attaching packages ───────────────────────────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.0 ✔ purrr 0.3.1
✔ tibble 2.0.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.4.0
✔ readr 1.3.1 ✔ forcats 0.4.0 Warning: package 'tibble' was built under R version 3.5.2Warning: package 'tidyr' was built under R version 3.5.2Warning: package 'purrr' was built under R version 3.5.2Warning: package 'dplyr' was built under R version 3.5.2Warning: package 'stringr' was built under R version 3.5.2Warning: package 'forcats' was built under R version 3.5.2── Conflicts ──────────────────────────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(reshape2)
Attaching package: 'reshape2'The following object is masked from 'package:tidyr':
smithslibrary(cowplot)Warning: package 'cowplot' was built under R version 3.5.2
Attaching package: 'cowplot'The following object is masked from 'package:ggplot2':
ggsaveTo overlap QTLs starting with eQTLs,pQTLs, ect and looking in the apaQTLs, I need to create empirical distributions to test as the null because the null is no longer uniform when there are genes with multiple peaks.
I need to match for number of peaks. To do this I should make a file with the nominal apaQTL genes and how many peaks were tested. I can make it a dictionary with the number of peaks then a list of genes with that number. that way i can randomly chose a matched when i go through the actual genes.
Question now: Pick a snp matched distance from the gene? How do I randomize which snps to choose?
For each actual QTL, i have to have the distance between the gene and the snp. I can use this to chose the snp for the empirical dist. Problem will be if snp is not tested in the set because it is over 1mb from the start of the peak.
nominal APA res are in: /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/
I can look at all of the snps tessted and see how many of the e/p qtls are not in that file:
eQTLs.
eQTLs=read.table("../data/other_qtls/fastqtl_qqnorm_RNAseq_phase2.fixed.perm.out",header=F, stringsAsFactors = F, col.names = c("pid", "nvar", "shape1", "shape2", "dummy", "sid", "dist", "npval", "ppval", "bpval", "bh")) %>% filter(-log10(bh)>1) pQTLs
pQTLs=read.table("../data/other_qtls/fastqtl_qqnorm_prot.fixed.perm.out",header=F, stringsAsFactors = F, col.names = c("pid", "nvar", "shape1", "shape2", "dummy", "sid", "dist", "npval", "ppval", "bpval", "bh")) %>% filter(-log10(bh)>1)Get all of the snps tested from the nominal apa res:
filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt
getTestedSnps.py
#python
apaRes=open("/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt","r")
testedsnps=open("/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/testedSnps.txt", "w")
snp_dic={}
for ln in apaRes:
snp=ln.split()[1]
if snp not in snp_dic.keys():
snp_dic[snp]=""
else:
continue
for each in snp_dic.keys():
testedsnps.write("%s\n"%(each))
testedsnps.close()
I want to add to this script to ask how many of the eQTL or pQTL snps are not in this list:
neqtlpqtlnottested.py
import numpy as np
apaRes=open("/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt","r")
eQTL=open("/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/perm/fastqtl_qqnorm_RNAseq_phase2.fixed.perm.out", "r")
pQTL=open("/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/perm/fastqtl_qqnorm_prot.fixed.perm.out", "r")
snpsNotTested= open("/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/eqltandpqtlSNPsnottestedinAPA.txt", "w")
snp_dic={}
for ln in apaRes:
snp=ln.split()[1]
if snp not in snp_dic.keys():
snp_dic[snp]=""
else:
continue
neqtl_not=0
neqtl=0
for ln in eQTL:
bh=ln.split()[10]
bh=float(bh)
if (np.log10(bh) * -1) > 1:
neqtl += 1
snp=ln.split()[5]
if snp not in snp_dic.keys():
neqtl_not +=1
npqtl=0
npqtl_not=0
for ln in pQTL:
bh=ln.split()[10]
bh=float(bh)
if (np.log10(bh) * -1) > 1:
npqtl +=1
snp=ln.split()[5]
if snp not in snp_dic.keys():
npqtl_not +=1
snpsNotTested.write("there are %s eQTLs and %s eQTLs not tested \n there are %s pQTLs and %s pQTLs not tested"%(neqtl,neqtl_not, npqtl,npqtl_not))
snpsNotTested.close()Results:
- there are 2438 eQTLs and 4 eQTLs not tested
- there are 674 pQTLs and 2 pQTLs not tested
This is not a huge number. I think we can ignore these for the current analysis.
Question now: Which snp.
I will pick the snp with the lowest pvalue when you look at the association in eQTL for that gene.
Step 1: fix names for eQTL files:
Eqtl files:
Perm res: /project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/fastqtl_qqnorm_RNAseq_phase2.fixed.perm.out
Now res: /project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/fastqtl_qqnorm_RNAseq_phase2.fixed.nominal.out
Annotation data= /project2/gilad/briana/genome_anotation_data/ensemble_to_genename.txt
Make file with sig eQTLs:
Run this interactviely in R
library(dplyr)
names=c('pid', 'nvar', 'shape1', 'shape2','dummy', 'sid', 'dist', 'npval' ,'slope', 'ppval', 'bpval' )
permRes=read.table("/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/fastqtl_qqnorm_RNAseq_phase2.fixed.perm.out",stringsAsFactors = F, col.names = names)
permRes$bh=p.adjust(permRes$bpval, method="fdr")
permRes_sig=permRes %>% filter(-log10(bh)>=1)
nonSig=permRes %>% filter(-log10(bh)<1)
write.table(permRes_sig, file="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_significanteQTLs.txt", col.names=F, row.names=F, quote=F, sep="\t")
write.table(nonSig, file="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_NOTeQTLs.txt", col.names=F, row.names=F, quote=F, sep="\t")Make a script to switch the gene names:
changeENSG2GeneName.py
def main(symbolF, genenameF):
nameFile=open("/project2/gilad/briana/genome_anotation_data/ensemble_to_genename.txt","r")
name_dic={}
for i,ln in enumerate(nameFile):
if i >0:
id, name, source =ln.split("\t")
name_dic[id]=name
inFile=open(symbolF, "r")
outFile=open(genenameF, "w")
nnoGene=0
for ln in inFile:
geneID=ln.split()[0].split(".")[0]
#print(geneID)
if geneID in name_dic.keys():
geneName=name_dic[geneID]
else:
nnoGene+=1
continue
otherStuff=ln.split()[1:]
line=[geneName, "\t".join(otherStuff)]
outString="\t".join(line)
#print(outString)
outFile.write("%s\n"%(outString))
print("%s Genes have no associated name in annoation"%(nnoGene))
outFile.close()
if __name__ == "__main__":
import sys
ensg=sys.argv[1]
geneName=sys.argv[2]
main(ensg, geneName)
Run for each file:
python changeENSG2GeneName.py /project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/fastqtl_qqnorm_RNAseq_phase2.fixed.perm.out /project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/fastqtl_qqnorm_RNAseq_phase2.fixed.perm_GeneNames.out
python changeENSG2GeneName.py /project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/fastqtl_qqnorm_RNAseq_phase2.fixed.nominal.out /project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/fastqtl_qqnorm_RNAseq_phase2.fixed.nominal_GeneNames.out
python changeENSG2GeneName.py /project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_significanteQTLs.txt /project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_significanteQTLs_GeneNames.txt
python changeENSG2GeneName.py /project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_NOTeQTLs.txt /project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_NOTeQTLs_eneNames.txtStep 2: Subset for tested in APA
Subset for genes tested in apa:
I need to subset the non eQTL genes for those actually tested in APA. This is important for the base distribution when I pick a random snp. I will have to do this seperate for total and nuclear similar to how i will have to look at number of peaks tested seperatly in total and nuclear. I will make a dictionary with the genes in the apa analysis, I can then go through the noneqtl file and keep only the genes with a gene in the dictionary.
subsetEQTL_testinAPA.py
totAPA="/project2/gilad/briana/threeprimeseq/data/perm_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Total.fixed.pheno_5perc_permRes.txt"
NucAPA="/project2/gilad/briana/threeprimeseq/data/perm_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Nuclear.fixed.pheno_5perc_permRes.txt"
nonEQTLs="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_NOTeQTLs_GeneNames.txt"
eQTLs="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_significanteQTLs_GeneNames.txt"
eQTLS_intot="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_eQTLs_GeneNames_inTot.txt"
eQTLS_innuc="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_eQTLs_GeneNames_inNuc.txt"
nonEQTL_intot="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_NOTeQTLs_GeneNames_inTot.txt"
nonEQTL_innuc="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_NOTeQTLs_GeneNames_inNuc.txt"
def check_names(inApa, ineqtl, outFile):
gene_dic={}
for ln in open(inApa, "r"):
gene=ln.split()[0].split(":")[-1].split("_")[0]
if gene not in gene_dic.keys():
gene_dic[gene]=""
else:
continue
fileOut= open(outFile, "w")
nnottested=0
for ln in open(ineqtl, "r"):
gene=ln.split()[0]
if gene in gene_dic.keys():
fileOut.write(ln)
else:
nnottested += 1
print("Number of genes not tested in apa = %s"%(nnottested))
fileOut.close()
print("total")
check_names(totAPA,nonEQTLs, nonEQTL_intot)
print("nuclear")
check_names(NucAPA,nonEQTLs, nonEQTL_innuc)
print("total")
check_names(totAPA,eQTLs, eQTLS_intot)
print("nuclear")
check_names(NucAPA,eQTLs, eQTLS_innuc)
There are about 4k genes of the 13k not tested.
Step 3: Make a file with # of peaks per gene:
I can make this in R with group by in the permuted data. I want a file that has the gene and how many peaks are in it.
TotPeaks=read.table("../data/perm_QTL_GeneLocAnno_noMP_5percov/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Total.fixed.pheno_5perc_permResBH.txt", header = T, stringsAsFactors = F) %>% select(pid) %>% separate(pid, into=c("chr", "start", "end", "peak"), sep=":") %>% separate(peak, into=c("gene", 'strand', 'peaknum'), sep="_") %>% group_by(gene) %>% summarise(NPeaks=n())Warning: Expected 3 pieces. Additional pieces discarded in 3 rows [886,
887, 888].write.table(TotPeaks, file="../data/EmpiricalDists/TotalQTL_nPeaks.txt", quote=F, sep="\t", col.names = F, row.names = F)
NucPeaks=read.table("../data/perm_QTL_GeneLocAnno_noMP_5percov/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Nuclear.fixed.pheno_5perc_permResBH.txt", header = T, stringsAsFactors = F) %>% select(pid) %>% separate(pid, into=c("chr", "start", "end", "peak"), sep=":") %>% separate(peak, into=c("gene", 'strand', 'peaknum'), sep="_") %>% group_by(gene) %>% summarise(NPeaks=n())Warning: Expected 3 pieces. Additional pieces discarded in 3 rows [1056,
1057, 1058].write.table(NucPeaks, file="../data/EmpiricalDists/NuclearQTL_nPeaks.txt", quote=F, sep="\t", col.names = F, row.names = F)Transfer to /project2/gilad/briana/threeprimeseq/data/EmpiricalDists/
Step 4 Make dist
makeeQTLEmpirical.py
def main(peaks, APA,nonE,eqtl, outF):
#dictionary of non egenes
nonEgenes={}
for ln in open(nonE):
gene=ln.split()[0]
nonEgenes[gene]=""
# make dictionary- key= npeak, value=list of genes with same value (use this when i need to pull random from the values)
#only add genes in the non eqet
peaknum={}
for ln in open(peaks, "r"):
gene, npeak = ln.split()
if npeak not in peaknum.keys():
if gene in nonEgenes.keys():
peaknum[npeak]=[gene]
else:
if gene in nonEgenes.keys():
peaknum[npeak].append(gene)
#make opposite dic- key =gene, value = npeaks (use this when i first get an EGENE to see how many peaks i will need)
#want all genes- e and no e genes
GenePeak={}
for ln in open(peaks, "r"):
gene, npeak= ln.split()
GenePeak[gene]=npeak
eQTLs= open(eqtl, "r")
for ln in eQTLs:
gene= ln.split()[0]
peaks2use=GenePeak[gene]
posibleGenes=peaknum[peaks2use]
if len(posibleGenes) == 0:
posibleGenes=peaknum[str(int(peaks2use)+1)]
if len(posibleGenes) == 0:
print("No genes with peaknum")
continue
GeneUse=random.choice(posibleGenes)
print(GeneUse)
#find the snp that goes with this gene in the non egenes file- this gives me the lowest pval snp
for ln in open(nonE,"r"):
genenon=ln.split()[0]
#print(genenon)
if genenon == GeneUse:
snpUse=ln.split()[5]
print(snpUse)
#now i have the gene and the snp, i can write out any of the nominal values from the apa file with these
fout=open(outF,"w")
for ln in open(APA, "r"):
geneApa=ln.split()[0].split(":")[-1].split("_")[0]
snpApa =ln.split()[1]
if geneApa==GeneUse and snpApa==snpUse:
fout.write(ln)
fout.close()
if __name__ == "__main__":
import sys
import random
fraction=sys.argv[1]
OutFile=sys.argv[2]
if fraction=="Total":
npeaks="/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/TotalQTL_nPeaks.txt"
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
nonEfile="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_NOTeQTLs_GeneNames_inTot.txt"
eQTL="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_eQTLs_GeneNames_inTot.txt"
else:
npeaks="/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/NuclearQTL_nPeaks.txt"
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
nonEfile="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_NOTeQTLs_GeneNames_inNuc.txt"
eQTL="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_eQTLs_GeneNames_inNuc.txt"
main(npeaks, nomFile, nonEfile, eQTL, OutFile)
python test_makeeQTLEmpirical.py Total "../data/EmpiricalDists/testTotal.txt"run_makeeQTLEmpirical.sh
#!/bin/bash
#SBATCH --job-name=run_makeeQTLEmpirical
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=run_makeeQTLEmpirical.out
#SBATCH --error=run_makeeQTLEmpirical.err
#SBATCH --partition=broadwl
#SBATCH --mem=36G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
python makeeQTLEmpirical.py "Total" "/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/eQTL_total_EmpiricalDist.txt"
python makeeQTLEmpirical.py "Nuclear" "/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/eQTL_nuclear_EmpiricalDist.txt"Overlap the eQTLs and apavalues
I want me make a script that takes in the total and nuclear eqtl overlap and nominal pvalues for the true dists.
getApaPval4eQTLs.py
def main(apa, eqtl, outFile):
fout=open(outFile,"w")
for ln in open(eQTL,"r"):
egene=ln.split()[0]
eSNP=ln.split()[5]
for ln in open(apa, "r"):
geneApa=ln.split()[0].split(":")[-1].split("_")[0]
snpApa =ln.split()[1]
if geneApa==egene and snpApa==eSNP:
fout.write(ln)
fout.close()
if __name__ == "__main__":
import sys
import random
fraction=sys.argv[1]
OutFile=sys.argv[2]
if fraction=="Total":
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
eQTL="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_eQTLs_GeneNames_inTot.txt"
else:
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
eQTL="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_eQTLs_GeneNames_inNuc.txt"
main(nomFile, eQTL, OutFile)
run_getApaPval4eQTLs.sh
#!/bin/bash
#SBATCH --job-name=run_getApaPval4eQTLs
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=run_getApaPval4eQTLs.out
#SBATCH --error=run_getApaPval4eQTLs.err
#SBATCH --partition=broadwl
#SBATCH --mem=36G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
python getApaPval4eQTLs.py "Total" "/project2/gilad/briana/threeprimeseq/data/eQTL_inAPA/eQTLinTotalApa.txt"
python getApaPval4eQTLs.py "Nuclear" "/project2/gilad/briana/threeprimeseq/data/eQTL_inAPA/eQTLinNuclearApa.txt"FASTER SCRIPTS:
getApaPval4eQTLs_secondtry.py
def main(apa, eqtl, outFile):
fout=open(outFile,"w")
use_dic={}
for ln in open(eQTL,"r"):
egene=ln.split()[0]
eSNP=ln.split()[5]
use_dic[egene]=eSNP
for ln in open(apa, "r"):
geneApa=ln.split()[0].split(":")[-1].split("_")[0]
snpApa =ln.split()[1]
if geneApa in use_dic.keys():
if snpApa == use_dic[geneApa]:
fout.write(ln)
fout.close()
if __name__ == "__main__":
import sys
import random
fraction=sys.argv[1]
OutFile=sys.argv[2]
if fraction=="Total":
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
eQTL="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_eQTLs_GeneNames_inTot.txt"
else:
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
eQTL="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_eQTLs_GeneNames_inNuc.txt"
main(nomFile, eQTL, OutFile)
run_getApaPval4eQTLs_second.sh
#!/bin/bash
#SBATCH --job-name=run_getApaPval4eQTLs2
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=run_getApaPval4eQTLs2.out
#SBATCH --error=run_getApaPval4eQTLs2.err
#SBATCH --partition=broadwl
#SBATCH --mem=36G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
python getApaPval4eQTLs_secondtry.py "Total" "/project2/gilad/briana/threeprimeseq/data/eQTL_inAPA/eQTLinTotalApa_try2.txt"
python getApaPval4eQTLs_secondtry.py "Nuclear" "/project2/gilad/briana/threeprimeseq/data/eQTL_inAPA/eQTLinNuclearApa_try2.txt"makeeQTLEmpirical_secondtry.py
def main(peaks, APA,nonE,eqtl, outF):
#dictionary of non egenes
nonEgenes={}
for ln in open(nonE):
gene=ln.split()[0]
nonEgenes[gene]=""
# make dictionary- key= npeak, value=list of genes with same value (use this when i need to pull random from the values)
#only add genes in the non eqet
peaknum={}
for ln in open(peaks, "r"):
gene, npeak = ln.split()
if npeak not in peaknum.keys():
if gene in nonEgenes.keys():
peaknum[npeak]=[gene]
else:
if gene in nonEgenes.keys():
peaknum[npeak].append(gene)
#make opposite dic- key =gene, value = npeaks (use this when i first get an EGENE to see how many peaks i will need)
#want all genes- e and no e genes
GenePeak={}
for ln in open(peaks, "r"):
gene, npeak= ln.split()
GenePeak[gene]=npeak
eQTLs= open(eqtl, "r")
gene_useDic={}
for ln in eQTLs:
gene= ln.split()[0]
peaks2use=GenePeak[gene]
posibleGenes=peaknum[peaks2use]
if len(posibleGenes) == 0:
print("No genes with peaknum")
posibleGenes=peaknum[str(int(peaks2use)+1)]
if len(posibleGenes) == 0:
print("No genes with peaknum")
continue
GeneUse=random.choice(posibleGenes)
gene_useDic[GeneUse]=""
#print(GeneUse)
#find the snp that goes with this gene in the non egenes file- this gives me the lowest pval snp
for ln in open(nonE,"r"):
genenon=ln.split()[0]
#print(genenon)
if genenon in gene_useDic.keys():
snpUse=ln.split()[5]
gene_useDic[genenon]=snpUse
#now i have the gene and the snp, i can write out any of the nominal values from the apa file with these
fout=open(outF,"w")
for ln in open(APA, "r"):
geneApa=ln.split()[0].split(":")[-1].split("_")[0]
snpApa =ln.split()[1]
if geneApa in gene_useDic.keys():
if snpApa == gene_useDic[geneApa]:
fout.write(ln)
fout.close()
if __name__ == "__main__":
import sys
import random
fraction=sys.argv[1]
OutFile=sys.argv[2]
if fraction=="Total":
npeaks="/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/TotalQTL_nPeaks.txt"
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
nonEfile="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_NOTeQTLs_GeneNames_inTot.txt"
eQTL="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_eQTLs_GeneNames_inTot.txt"
else:
npeaks="/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/NuclearQTL_nPeaks.txt"
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
nonEfile="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_NOTeQTLs_GeneNames_inNuc.txt"
eQTL="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_eQTLs_GeneNames_inNuc.txt"
main(npeaks, nomFile, nonEfile, eQTL, OutFile)
run_makeeQTLEmpirical_secondtry.sh
#!/bin/bash
#SBATCH --job-name=run_makeeQTLEmpirical2
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=run_makeeQTLEmpirical2.out
#SBATCH --error=run_makeeQTLEmpirical2.err
#SBATCH --partition=broadwl
#SBATCH --mem=36G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
python makeeQTLEmpirical_secondtry.py "Total" "/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/eQTL_total_EmpiricalDist_secondtry.txt"
python makeeQTLEmpirical_secondtry.py "Nuclear" "/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/eQTL_nuclear_EmpiricalDist_secondtry.txt"QQ plots
qqplot(-log10(empiricalPval), -log10(actualPval))
Actual Dists:
nomnames=c("peakID", 'snp','dist', 'pval', 'slope')
eQTLinTotal=read.table("../data/eQTL_inAPA/eQTLinTotalApa_try2.txt", stringsAsFactors = F, col.names = nomnames)
eQTLinNuclear=read.table("../data/eQTL_inAPA/eQTLinNuclearApa_try2.txt", stringsAsFactors = F, col.names = nomnames)Emiprical:
empTotal=read.table("../data/EmpiricalDists/eQTL_total_EmpiricalDist_secondtry.txt", col.names = nomnames,stringsAsFactors = F)
empNuclear=read.table("../data/EmpiricalDists/eQTL_nuclear_EmpiricalDist_secondtry.txt", col.names = nomnames, stringsAsFactors = F)Deal with problem that there are less snps in empirical (add some uniform)
toaddTotal=runif(nrow(eQTLinTotal)-nrow(empTotal))
toaddNuclear=runif(nrow(eQTLinNuclear)-nrow(empNuclear))Add the extra pvalues:
empNuclearUse= c(as.vector(empNuclear$pval),toaddNuclear)
empTotalUse= c(as.vector(empTotal$pval),toaddTotal)make total plot:
png("../output/plots/eqtlsinTotAPAQQPlot_ALLeqtl.png")
qqplot(-log10(empTotalUse), -log10(eQTLinTotal$pval),ylab="-log10 Total APA pval", xlab="Empirical expectation", main="eQTLs in totalAPA analysis")
abline(0,1)
dev.off()quartz_off_screen
2 make nuclear:
png("../output/plots/eqtlsinNucAPAQQPlot_ALLeqtl.png")
qqplot(-log10(empNuclearUse), -log10(eQTLinTotal$pval),ylab="-log10 Nuclear APA pval", xlab="Empirical expectation", main="eQTLs in nuclearAPA analysis")
abline(0,1)
dev.off()quartz_off_screen
2 I will want to subset the actual to just get the unexplained ones:
/project2/gilad/briana/threeprimeseq/data/eQTL_Lietal/unexplained_FDR10.sort.txt
This has the unexplained eQTLs, I need to get all of the genes and switch symbol to gene name.
fixUnexplainedNames.py
geneNames=open("/project2/gilad/briana/genome_anotation_data/ensemble_to_genename.txt","r")
name_dic={}
for i,ln in enumerate(geneNames):
if i >0:
id, name, source =ln.split("\t")
name_dic[id]=name
inFile=open("/project2/gilad/briana/threeprimeseq/data/eQTL_Lietal/unexplained_FDR10.sort.txt","r")
outFile=open("/project2/gilad/briana/threeprimeseq/data/eQTL_Lietal/unexplained_FDR10_GENEnames.txt", "w")
nNot=0
for ln in inFile:
line_list=ln.split()
if len(line_list)==3:
geneid=ln.split()[2]
if geneid in name_dic.keys():
genename=name_dic[geneid]
outFile.write("%s\n"%(genename))
else:
nNot+=1
else:
geneList=line_list[2:]
for each in geneList:
if each in name_dic.keys():
genename=name_dic[each]
outFile.write("%s\n"%(genename))
else:
nNot+=1
print(nNot)
outFile.close()
Subset eQTLs for these unexplained eQTL gene.
subseteqtlUnexp.py
eQTLS_intot="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_eQTLs_GeneNames_inTot.txt"
Unexp_eQTLS_intot="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_Unexplained_eQTLs_GeneNames_inTot.txt"
eQTLS_innuc="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_eQTLs_GeneNames_inNuc.txt"
Unexp_eQTLS_innuc="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_Unexplained_eQTLs_GeneNames_inNuc.txt"
def subsetQTL(inQTL, unexQTL):
ungenes={}
for ln in open("/project2/gilad/briana/threeprimeseq/data/eQTL_Lietal/unexplained_FDR10_GENEnames.txt","r"):
gene=ln.strip()
ungenes[gene]=""
fout=open(unexQTL, "w")
for ln in open(inQTL, "r"):
egene=ln.split()[0]
if egene in ungenes.keys():
fout.write(ln)
fout.close()
subsetQTL(eQTLS_intot,Unexp_eQTLS_intot)
subsetQTL(eQTLS_innuc,Unexp_eQTLS_innuc)
Get read and empirical for these:
getApaPval4eQTLs_secondtry_UNEXP.py
def main(apa, eqtl, outFile):
fout=open(outFile,"w")
use_dic={}
for ln in open(eQTL,"r"):
egene=ln.split()[0]
eSNP=ln.split()[5]
use_dic[egene]=eSNP
for ln in open(apa, "r"):
geneApa=ln.split()[0].split(":")[-1].split("_")[0]
snpApa =ln.split()[1]
if geneApa in use_dic.keys():
if snpApa == use_dic[geneApa]:
fout.write(ln)
fout.close()
if __name__ == "__main__":
import sys
import random
fraction=sys.argv[1]
OutFile=sys.argv[2]
if fraction=="Total":
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
eQTL="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_Unexplained_eQTLs_GeneNames_inTot.txt"
else:
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
eQTL="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_Unexplained_eQTLs_GeneNames_inNuc.txt"
main(nomFile, eQTL, OutFile)
run_getApaPval4eQTLs_second_unexp.sh
#!/bin/bash
#SBATCH --job-name=run_getApaPval4eQTLs2_unex
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=run_getApaPval4eQTLs2_unex.out
#SBATCH --error=run_getApaPval4eQTLs2_unex.err
#SBATCH --partition=broadwl
#SBATCH --mem=36G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
python getApaPval4eQTLs_secondtry_UNEXP.py "Total" "/project2/gilad/briana/threeprimeseq/data/eQTL_inAPA/UnexpeQTLinTotalApa_try2.txt"
python getApaPval4eQTLs_secondtry_UNEXP.py "Nuclear" "/project2/gilad/briana/threeprimeseq/data/eQTL_inAPA/UnexpeQTLinNuclearApa_try2.txt"makeeQTLEmpirical_secondtry_unex.py
def main(peaks, APA,nonE,eqtl, outF):
#dictionary of non egenes
nonEgenes={}
for ln in open(nonE):
gene=ln.split()[0]
nonEgenes[gene]=""
# make dictionary- key= npeak, value=list of genes with same value (use this when i need to pull random from the values)
#only add genes in the non eqet
peaknum={}
for ln in open(peaks, "r"):
gene, npeak = ln.split()
if npeak not in peaknum.keys():
if gene in nonEgenes.keys():
peaknum[npeak]=[gene]
else:
if gene in nonEgenes.keys():
peaknum[npeak].append(gene)
#make opposite dic- key =gene, value = npeaks (use this when i first get an EGENE to see how many peaks i will need)
#want all genes- e and no e genes
GenePeak={}
for ln in open(peaks, "r"):
gene, npeak= ln.split()
GenePeak[gene]=npeak
eQTLs= open(eqtl, "r")
gene_useDic={}
for ln in eQTLs:
gene= ln.split()[0]
peaks2use=GenePeak[gene]
posibleGenes=peaknum[peaks2use]
if len(posibleGenes) == 0:
print("No genes with peaknum")
posibleGenes=peaknum[str(int(peaks2use)+1)]
if len(posibleGenes) == 0:
print("No genes with peaknum")
continue
GeneUse=random.choice(posibleGenes)
gene_useDic[GeneUse]=""
#print(GeneUse)
#find the snp that goes with this gene in the non egenes file- this gives me the lowest pval snp
for ln in open(nonE,"r"):
genenon=ln.split()[0]
#print(genenon)
if genenon in gene_useDic.keys():
snpUse=ln.split()[5]
gene_useDic[genenon]=snpUse
#now i have the gene and the snp, i can write out any of the nominal values from the apa file with these
fout=open(outF,"w")
for ln in open(APA, "r"):
geneApa=ln.split()[0].split(":")[-1].split("_")[0]
snpApa =ln.split()[1]
if geneApa in gene_useDic.keys():
if snpApa == gene_useDic[geneApa]:
fout.write(ln)
fout.close()
if __name__ == "__main__":
import sys
import random
fraction=sys.argv[1]
OutFile=sys.argv[2]
if fraction=="Total":
npeaks="/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/TotalQTL_nPeaks.txt"
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
nonEfile="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_NOTeQTLs_GeneNames_inTot.txt"
eQTL="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_Unexplained_eQTLs_GeneNames_inTot.txt"
else:
npeaks="/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/NuclearQTL_nPeaks.txt"
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
nonEfile="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_NOTeQTLs_GeneNames_inNuc.txt"
eQTL="/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_Unexplained_eQTLs_GeneNames_inNuc.txt"
main(npeaks, nomFile, nonEfile, eQTL, OutFile)
run_makeeQTLEmpirical_secondtry_unexp.sh
#!/bin/bash
#SBATCH --job-name=run_makeeQTLEmpirical2_unexp
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=run_makeeQTLEmpirical2_unexp.out
#SBATCH --error=run_makeeQTLEmpirical2_unexp.err
#SBATCH --partition=broadwl
#SBATCH --mem=36G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
python makeeQTLEmpirical_secondtry_unex.py "Total" "/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/eQTL_total_EmpiricalDist_secondtry_UNEXP.txt"
python makeeQTLEmpirical_secondtry_unex.py "Nuclear" "/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/eQTL_nuclear_EmpiricalDist_secondtry_UNEXP.txt"points(sort(-log10(runif(nrow(t)))), sort(-log10(t$corrPval)), col= alpha(“Red”)) abline(0,1)
UneQTLinTotal=read.table("../data/eQTL_inAPA/UnexpeQTLinTotalApa_try2.txt", stringsAsFactors = F, col.names = nomnames)
UNeQTLinNuclear=read.table("../data/eQTL_inAPA/UnexpeQTLinNuclearApa_try2.txt", stringsAsFactors = F, col.names = nomnames)Emiprical:
empTotalUn=read.table("../data/EmpiricalDists/eQTL_total_EmpiricalDist_secondtry_UNEXP.txt", col.names = nomnames,stringsAsFactors = F)
empNuclearUn=read.table("../data/EmpiricalDists/eQTL_nuclear_EmpiricalDist_secondtry_UNEXP.txt", col.names = nomnames, stringsAsFactors = F)Deal with problem that there are less snps in empirical (add some uniform)
toaddTotalUn=runif(nrow(UneQTLinTotal)-nrow(empTotalUn))
toaddNuclearUn=runif(nrow(UNeQTLinNuclear)-nrow(empNuclearUn))Add the extra pvalues:
empNuclearUseUN= c(as.vector(empNuclearUn$pval),toaddNuclearUn)
empTotalUseUN= c(as.vector(empTotalUn$pval),toaddTotalUn)make total plot:
png("../output/plots/eqtlsinTotAPAQQPlot.png")
qqplot(-log10(empTotalUse), -log10(eQTLinTotal$pval),ylab="-log10 Total APA pval", xlab="Empirical expectation", main="eQTLs in totalAPA analysis")
points(sort(-log10(empTotalUseUN)), sort(-log10(UneQTLinTotal$pval)),col= alpha("Red"))
legend("topleft", legend=c("All eQTLs", "Unexplained eQTLs"),col=c("black", "red"), pch=16,bty = 'n')
abline(0,1)
dev.off()quartz_off_screen
2 Nuclear plot:
png("../output/plots/eqtlsinNucAPAQQPlot.png")
qqplot(-log10(empNuclearUse), -log10(eQTLinNuclear$pval),ylab="-log10 Nuclear APA pval", xlab="Empirical expectation", main="eQTLs in nuclearAPA analysis")
points(sort(-log10(empNuclearUseUN)), sort(-log10(UNeQTLinNuclear$pval)),col= alpha("Red"))
legend("topleft", legend=c("All eQTLs", "Unexplained eQTLs"),col=c("black", "red"), pch=16,bty = 'n')
abline(0,1)
dev.off()quartz_off_screen
2 Same analysis starting with pQTLs
Make file with sig pQTLs:
Run this interactviely in R
library(dplyr)
names=c('pid', 'nvar', 'shape1', 'shape2','dummy', 'sid', 'dist', 'npval' ,'slope', 'ppval', 'bpval' )
permRes=read.table("/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/fastqtl_qqnorm_prot.fixed.perm.out",stringsAsFactors = F, col.names = names)
permRes$bh=p.adjust(permRes$bpval, method="fdr")
permRes_sig=permRes %>% filter(-log10(bh)>=1)
nonSig=permRes %>% filter(-log10(bh)<1)
write.table(permRes_sig, file="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_significantpQTLs.txt", col.names=F, row.names=F, quote=F, sep="\t")
write.table(nonSig, file="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_NOTpQTLs.txt", col.names=F, row.names=F, quote=F, sep="\t")Run for each file:
python changeENSG2GeneName.py /project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_significantpQTLs.txt /project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_significantpQTLs_GeneNames.txt
python changeENSG2GeneName.py /project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_NOTpQTLs.txt /project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_NOTpQTLs_GeneNames.txt
python changeENSG2GeneName.py /project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/fastqtl_qqnorm_prot_fixed.nominal.out /project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/fastqtl_qqnorm_prot_fixed.nominal_GeneNames.out
Subset genes that were tested in apa.
subsetpQTL_testinAPA.py
totAPA="/project2/gilad/briana/threeprimeseq/data/perm_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Total.fixed.pheno_5perc_permRes.txt"
NucAPA="/project2/gilad/briana/threeprimeseq/data/perm_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Nuclear.fixed.pheno_5perc_permRes.txt"
nonPQTLs="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_NOTpQTLs_GeneNames.txt"
pQTLs="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_significantpQTLs_GeneNames.txt"
pQTLS_intot="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_pQTLs_GeneNames_inTot.txt"
pQTLS_innuc="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_pQTLs_GeneNames_inNuc.txt"
nonPQTL_intot="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_NOTpQTLs_GeneNames_inTot.txt"
nonPQTL_innuc="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_NOTpQTLs_GeneNames_inNuc.txt"
def check_names(inApa, inqtl, outFile):
gene_dic={}
for ln in open(inApa, "r"):
gene=ln.split()[0].split(":")[-1].split("_")[0]
if gene not in gene_dic.keys():
gene_dic[gene]=""
else:
continue
fileOut= open(outFile, "w")
nnottested=0
for ln in open(inqtl, "r"):
gene=ln.split()[0]
if gene in gene_dic.keys():
fileOut.write(ln)
else:
nnottested += 1
print("Number of genes not tested in apa = %s"%(nnottested))
fileOut.close()
print("total")
check_names(totAPA,nonPQTLs, nonPQTL_intot)
print("nuclear")
check_names(NucAPA,nonPQTLs, nonPQTL_innuc)
print("total")
check_names(totAPA,pQTLs, pQTLS_intot)
print("nuclear")
check_names(NucAPA,pQTLs, pQTLS_innuc)non PQTLS:
total Number of genes not tested in apa = 473 nuclear Number of genes not tested in apa = 444
PQTLS:
total Number of genes not tested in apa = 8 nuclear Number of genes not tested in apa = 8
Make distributions:
getApaPval4pQTLs_secondtry.py
def main(apa, pqtl, outFile):
fout=open(outFile,"w")
use_dic={}
for ln in open(pQTL,"r"):
pgene=ln.split()[0]
pSNP=ln.split()[5]
use_dic[pgene]=pSNP
for ln in open(apa, "r"):
geneApa=ln.split()[0].split(":")[-1].split("_")[0]
snpApa =ln.split()[1]
if geneApa in use_dic.keys():
if snpApa == use_dic[geneApa]:
fout.write(ln)
fout.close()
if __name__ == "__main__":
import sys
import random
fraction=sys.argv[1]
OutFile=sys.argv[2]
if fraction=="Total":
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
pQTL="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_pQTLs_GeneNames_inTot.txt"
else:
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
pQTL="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_pQTLs_GeneNames_inNuc.txt"
main(nomFile, pQTL, OutFile)
run_getApaPval4pQTLs_second.sh
#!/bin/bash
#SBATCH --job-name=run_getApaPval4pQTLs2
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=run_getApaPval4pQTLs2.out
#SBATCH --error=run_getApaPval4pQTLs2.err
#SBATCH --partition=broadwl
#SBATCH --mem=36G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
python getApaPval4pQTLs_secondtry.py "Total" "/project2/gilad/briana/threeprimeseq/data/pQTL_inAPA/pQTLinTotalApa_try2.txt"
python getApaPval4pQTLs_secondtry.py "Nuclear" "/project2/gilad/briana/threeprimeseq/data/pQTL_inAPA/pQTLinNuclearApa_try2.txt"makepQTLEmpirical_secondtry.py
def main(peaks, APA,nonP,pqtl, outF):
#dictionary of non egenes
nonPgenes={}
for ln in open(nonP):
gene=ln.split()[0]
nonPgenes[gene]=""
# make dictionary- key= npeak, value=list of genes with same value (use this when i need to pull random from the values)
#only add genes in the non eqet
peaknum={}
for ln in open(peaks, "r"):
gene, npeak = ln.split()
if npeak not in peaknum.keys():
if gene in nonPgenes.keys():
peaknum[npeak]=[gene]
else:
if gene in nonPgenes.keys():
peaknum[npeak].append(gene)
#make opposite dic- key =gene, value = npeaks (use this when i first get an EGENE to see how many peaks i will need)
#want all genes- e and no e genes
GenePeak={}
for ln in open(peaks, "r"):
gene, npeak= ln.split()
GenePeak[gene]=npeak
pQTLs= open(pqtl, "r")
gene_useDic={}
for ln in pQTLs:
gene= ln.split()[0]
peaks2use=GenePeak[gene]
posibleGenes=peaknum[peaks2use]
if len(posibleGenes) == 0:
print("No genes with peaknum")
posibleGenes=peaknum[str(int(peaks2use)+1)]
if len(posibleGenes) == 0:
print("No genes with peaknum")
continue
GeneUse=random.choice(posibleGenes)
gene_useDic[GeneUse]=""
#print(GeneUse)
#find the snp that goes with this gene in the non egenes file- this gives me the lowest pval snp
for ln in open(nonP,"r"):
genenon=ln.split()[0]
#print(genenon)
if genenon in gene_useDic.keys():
snpUse=ln.split()[5]
gene_useDic[genenon]=snpUse
#now i have the gene and the snp, i can write out any of the nominal values from the apa file with these
fout=open(outF,"w")
for ln in open(APA, "r"):
geneApa=ln.split()[0].split(":")[-1].split("_")[0]
snpApa =ln.split()[1]
if geneApa in gene_useDic.keys():
if snpApa == gene_useDic[geneApa]:
fout.write(ln)
fout.close()
if __name__ == "__main__":
import sys
import random
fraction=sys.argv[1]
OutFile=sys.argv[2]
if fraction=="Total":
npeaks="/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/TotalQTL_nPeaks.txt"
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
nonPfile="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_NOTpQTLs_GeneNames_inTot.txt"
pQTL="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_pQTLs_GeneNames_inTot.txt"
else:
npeaks="/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/NuclearQTL_nPeaks.txt"
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
nonPfile="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_NOTpQTLs_GeneNames_inNuc.txt"
pQTL="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_pQTLs_GeneNames_inNuc.txt"
main(npeaks, nomFile, nonPfile, pQTL, OutFile) run_makepQTLEmpirical_secondtry.sh
#!/bin/bash
#SBATCH --job-name=run_makepQTLEmpirical2
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=run_makepQTLEmpirical2.out
#SBATCH --error=run_makepQTLEmpirical2.err
#SBATCH --partition=broadwl
#SBATCH --mem=36G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
python makepQTLEmpirical_secondtry.py "Total" "/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/pQTL_total_EmpiricalDist_secondtry.txt"
python makepQTLEmpirical_secondtry.py "Nuclear" "/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/pQTL_nuclear_EmpiricalDist_secondtry.txt"
pQTLinTotal=read.table("../data/pQTL_inAPA/pQTLinTotalApa_try2.txt", stringsAsFactors = F, col.names = nomnames)
pQTLinNuclear=read.table("../data/pQTL_inAPA/pQTLinNuclearApa_try2.txt", stringsAsFactors = F, col.names = nomnames)
PempTotal=read.table("../data/EmpiricalDists/pQTL_total_EmpiricalDist_secondtry.txt", col.names = nomnames,stringsAsFactors = F)
PempNuclear=read.table("../data/EmpiricalDists/pQTL_nuclear_EmpiricalDist_secondtry.txt", col.names = nomnames, stringsAsFactors = F)ptoaddTotal=runif(nrow(pQTLinTotal)-nrow(PempTotal))
PempTotalUse= c(as.vector(PempTotal$pval),ptoaddTotal)
#For the nuclear I need to randomly remove 1:
PempNuclearUse=PempNuclear %>% slice(1:130) png("../output/plots/pqtlsinTotAPAQQPlot.png")
qqplot(-log10(PempTotalUse), -log10(pQTLinTotal$pval),ylab="-log10 Total APA pval", xlab="Empirical expectation", main="pQTLs in totalAPA analysis")
abline(0,1)
dev.off()quartz_off_screen
2 png("../output/plots/pqtlsinNucAPAQQPlot.png")
qqplot(-log10(PempNuclearUse$pval), -log10(pQTLinNuclear$pval),ylab="-log10 Nuclear APA pval", xlab="Empirical expectation", main="pQTLs in nuclearAPA analysis")
abline(0,1)
dev.off()quartz_off_screen
2 Pqtls that are not eQTLs
subset for pgenes that are not e genes
subsetpQTLsnoteQTLs.py
#pqtl
totP="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_pQTLs_GeneNames_inTot.txt"
nucP="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_pQTLs_GeneNames_inNuc.txt"
totPnotE="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_pQTLs_GeneNames_inTot_notE.txt"
nucPnotE="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_pQTLs_GeneNames_inNuc_notE.txt"
def subsetP(pqtl, pnote):
#eQTL genes
eQTLs=open("/project2/gilad/briana/threeprimeseq/data/eQTL_myanalysis/permRes_significanteQTLs_GeneNames.txt","r")
egenes={}
for ln in eQTLs:
gene=ln.split()[0]
egenes[gene]=""
fout=open(pnote,"w")
for ln in open(pqtl,"r"):
gene=ln.split()[0]
if gene not in egenes.keys():
fout.write(ln)
fout.close()
subsetP(totP, totPnotE)
subsetP(nucP, nucPnotE)getApaPval4pQTLs_secondtry_notE.py
def main(apa, pqtl, outFile):
fout=open(outFile,"w")
use_dic={}
for ln in open(pQTL,"r"):
pgene=ln.split()[0]
pSNP=ln.split()[5]
use_dic[pgene]=pSNP
for ln in open(apa, "r"):
geneApa=ln.split()[0].split(":")[-1].split("_")[0]
snpApa =ln.split()[1]
if geneApa in use_dic.keys():
if snpApa == use_dic[geneApa]:
fout.write(ln)
fout.close()
if __name__ == "__main__":
import sys
import random
fraction=sys.argv[1]
OutFile=sys.argv[2]
if fraction=="Total":
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
pQTL="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_pQTLs_GeneNames_inTot_notE.txt"
else:
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
pQTL="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_pQTLs_GeneNames_inNuc_notE.txt"
main(nomFile, pQTL, OutFile)
run_getApaPval4pQTLs_second_notE.sh
#!/bin/bash
#SBATCH --job-name=run_getApaPval4pQTLs2_notE
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=run_getApaPval4pQTLs2_notE.out
#SBATCH --error=run_getApaPval4pQTLs2_notE.err
#SBATCH --partition=broadwl
#SBATCH --mem=36G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
python getApaPval4pQTLs_secondtry_notE.py "Total" "/project2/gilad/briana/threeprimeseq/data/pQTL_inAPA/pQTLinTotalApa_try2_notE.txt"
python getApaPval4pQTLs_secondtry_notE.py "Nuclear" "/project2/gilad/briana/threeprimeseq/data/pQTL_inAPA/pQTLinNuclearApa_try2_notE.txt"makepQTLEmpirical_secondtry_notE.py
def main(peaks, APA,nonP,pqtl, outF):
#dictionary of non egenes
nonPgenes={}
for ln in open(nonP):
gene=ln.split()[0]
nonPgenes[gene]=""
# make dictionary- key= npeak, value=list of genes with same value (use this when i need to pull random from the values)
#only add genes in the non eqet
peaknum={}
for ln in open(peaks, "r"):
gene, npeak = ln.split()
if npeak not in peaknum.keys():
if gene in nonPgenes.keys():
peaknum[npeak]=[gene]
else:
if gene in nonPgenes.keys():
peaknum[npeak].append(gene)
#make opposite dic- key =gene, value = npeaks (use this when i first get an EGENE to see how many peaks i will need)
#want all genes- e and no e genes
GenePeak={}
for ln in open(peaks, "r"):
gene, npeak= ln.split()
GenePeak[gene]=npeak
pQTLs= open(pqtl, "r")
gene_useDic={}
for ln in pQTLs:
gene= ln.split()[0]
peaks2use=GenePeak[gene]
posibleGenes=peaknum[peaks2use]
if len(posibleGenes) == 0:
print("No genes with peaknum")
posibleGenes=peaknum[str(int(peaks2use)+1)]
if len(posibleGenes) == 0:
print("No genes with peaknum")
continue
GeneUse=random.choice(posibleGenes)
gene_useDic[GeneUse]=""
#print(GeneUse)
#find the snp that goes with this gene in the non egenes file- this gives me the lowest pval snp
for ln in open(nonP,"r"):
genenon=ln.split()[0]
#print(genenon)
if genenon in gene_useDic.keys():
snpUse=ln.split()[5]
gene_useDic[genenon]=snpUse
#now i have the gene and the snp, i can write out any of the nominal values from the apa file with these
fout=open(outF,"w")
for ln in open(APA, "r"):
geneApa=ln.split()[0].split(":")[-1].split("_")[0]
snpApa =ln.split()[1]
if geneApa in gene_useDic.keys():
if snpApa == gene_useDic[geneApa]:
fout.write(ln)
fout.close()
if __name__ == "__main__":
import sys
import random
fraction=sys.argv[1]
OutFile=sys.argv[2]
if fraction=="Total":
npeaks="/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/TotalQTL_nPeaks.txt"
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Total.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
nonPfile="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_NOTpQTLs_GeneNames_inTot.txt"
pQTL="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_pQTLs_GeneNames_inTot_notE.txt"
else:
npeaks="/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/NuclearQTL_nPeaks.txt"
nomFile="/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_GeneLocAnno_noMP_5percUs/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_NoMP_sm_quant.Nuclear.fixed.pheno_5perc.fc.gz.qqnorm_allNomRes.txt"
nonPfile="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_NOTpQTLs_GeneNames_inNuc.txt"
pQTL="/project2/gilad/briana/threeprimeseq/data/pQTL_myanalysis/permRes_pQTLs_GeneNames_inNuc_notE.txt"
main(npeaks, nomFile, nonPfile, pQTL, OutFile) run_makepQTLEmpirical_secondtry_notE.sh
#!/bin/bash
#SBATCH --job-name=run_makepQTLEmpirical2_notE
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=run_makepQTLEmpirical2_notE.out
#SBATCH --error=run_makepQTLEmpirical2_notE.err
#SBATCH --partition=broadwl
#SBATCH --mem=36G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
python makepQTLEmpirical_secondtry_notE.py "Total" "/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/pQTL_total_EmpiricalDist_secondtry_notE.txt"
python makepQTLEmpirical_secondtry_notE.py "Nuclear" "/project2/gilad/briana/threeprimeseq/data/EmpiricalDists/pQTL_nuclear_EmpiricalDist_secondtry_notE.txt"
pQTLinTotal_notE=read.table("../data/pQTL_inAPA/pQTLinTotalApa_try2_notE.txt", stringsAsFactors = F, col.names = nomnames)
pQTLinNuclear_notE=read.table("../data/pQTL_inAPA/pQTLinNuclearApa_try2_notE.txt", stringsAsFactors = F, col.names = nomnames)PempTotalnotE=read.table("../data/EmpiricalDists/pQTL_total_EmpiricalDist_secondtry_notE.txt", col.names = nomnames,stringsAsFactors = F)
PempNuclearnotE=read.table("../data/EmpiricalDists/pQTL_nuclear_EmpiricalDist_secondtry_notE.txt", col.names = nomnames, stringsAsFactors = F)toaddTotalPnotE=runif(nrow(pQTLinTotal_notE)-nrow(PempTotalnotE))
toaddNuclearPnotE=runif(nrow(pQTLinNuclear_notE)-nrow(PempNuclearnotE))
PempNuclearUsenotE= c(as.vector(PempNuclearnotE$pval),toaddNuclearPnotE)
PempTotalUsenotE= c(as.vector(PempTotalnotE$pval),toaddTotalPnotE)png("../output/plots/pqtlsinTotAPAQQPlot_withnonE.png")
qqplot(-log10(PempTotalUse), -log10(pQTLinTotal$pval),ylab="-log10 Total APA pval", xlab="Empirical expectation", main="pQTLs in totalAPA analysis")
points(sort(-log10(PempTotalUsenotE)), sort(-log10(pQTLinTotal_notE$pval)),col=alpha("Red"))
legend("topleft", legend=c("All pQTLs", "non Egene pQTLs"),col=c("black", "red"), pch=16,bty = 'n')
abline(0,1)
dev.off()quartz_off_screen
2 png("../output/plots/pqtlsinNucAPAQQPlot_withnonE.png")
qqplot(-log10(PempNuclearUse$pval), -log10(pQTLinNuclear$pval),ylab="-log10 Nuclear APA pval", xlab="Empirical expectation", main="pQTLs in nuclearAPA analysis")
points(sort(-log10(PempNuclearUsenotE)), sort(-log10(pQTLinNuclear_notE$pval)),col=alpha("Red"))
legend("topleft", legend=c("All pQTLs", "non Egene pQTLs"),col=c("black", "red"), pch=16,bty = 'n')
abline(0,1)
dev.off()quartz_off_screen
2 Investigate top p not e
pQTLinTotal_notE %>% mutate(fixedPval=-log10(pval)) %>% arrange(desc(fixedPval)) %>% filter(fixedPval>2) peakID snp dist pval
1 22:36622669:36622752:APOL2_+_peak104256 22:36614412 -8258 6.65193e-07
2 2:191074768:191074850:HIBCH_+_peak93667 2:191148347 73578 1.51247e-04
3 8:87484579:87484661:RMDN1_+_peak160108 8:87515611 31031 2.24485e-03
4 22:36622255:36622339:APOL2_+_peak104255 22:36614412 -7844 3.92721e-03
5 8:87486452:87486566:RMDN1_+_peak160112 8:87515611 29158 4.13078e-03
6 1:233105742:233105826:NTPCR_-_peak15557 1:233113196 7453 4.14054e-03
slope fixedPval
1 -0.770876 6.177052
2 -0.679005 3.820313
3 0.820206 2.648813
4 0.564080 2.405916
5 -0.670974 2.383968
6 -0.296659 2.382943pQTLinNuclear_notE %>% mutate(fixedPval=-log10(pval)) %>% arrange(desc(fixedPval)) %>% filter(fixedPval>2) peakID snp dist pval
1 2:191074768:191074850:HIBCH_+_peak93667 2:191148347 73578 4.44406e-12
2 22:36622669:36622752:APOL2_+_peak104256 22:36614412 -8258 1.48711e-06
3 22:36622255:36622339:APOL2_+_peak104255 22:36614412 -7844 6.88375e-06
4 8:87484579:87484661:RMDN1_+_peak160108 8:87515611 31031 4.09927e-03
5 16:50134231:50134323:HEATR3_-_peak62683 16:50089070 -45162 6.05304e-03
6 2:191069361:191069449:HIBCH_+_peak93659 2:191148347 78985 8.61873e-03
slope fixedPval
1 -1.217860 11.352220
2 -0.783432 5.827657
3 0.741472 5.162175
4 0.850781 2.387293
5 -0.482404 2.218026
6 0.240519 2.064557Join these and look at the difference:
pQTLinTotal_notE_format= pQTLinTotal_notE %>% mutate(TotalfixedPval=-log10(pval)) %>% select(peakID, snp, TotalfixedPval)
pQTLinNuclear_notE_format= pQTLinNuclear_notE %>% mutate(NuclearfixedPval=-log10(pval)) %>% select(peakID, snp, NuclearfixedPval)
pQTLBoth_notE=pQTLinTotal_notE_format %>% inner_join(pQTLinNuclear_notE_format, by=c("peakID", "snp")) %>% separate(peakID, into=c("chr", "start", "end", "peak"), sep=":") %>% separate(peak, into = c("gene", "strand", "peakNum"),sep="_")Plot
ggplot(pQTLBoth_notE,aes(x=TotalfixedPval,y=NuclearfixedPval)) + geom_point() + geom_abline(slope=1) + labs(x="-log10(Total Pval)",y="-log10(Nuclear Pval)", title="apa association pvalue for pQTLs in non E genes") + geom_text(aes(label=gene),nudge_x = .35) + geom_vline(xintercept =1)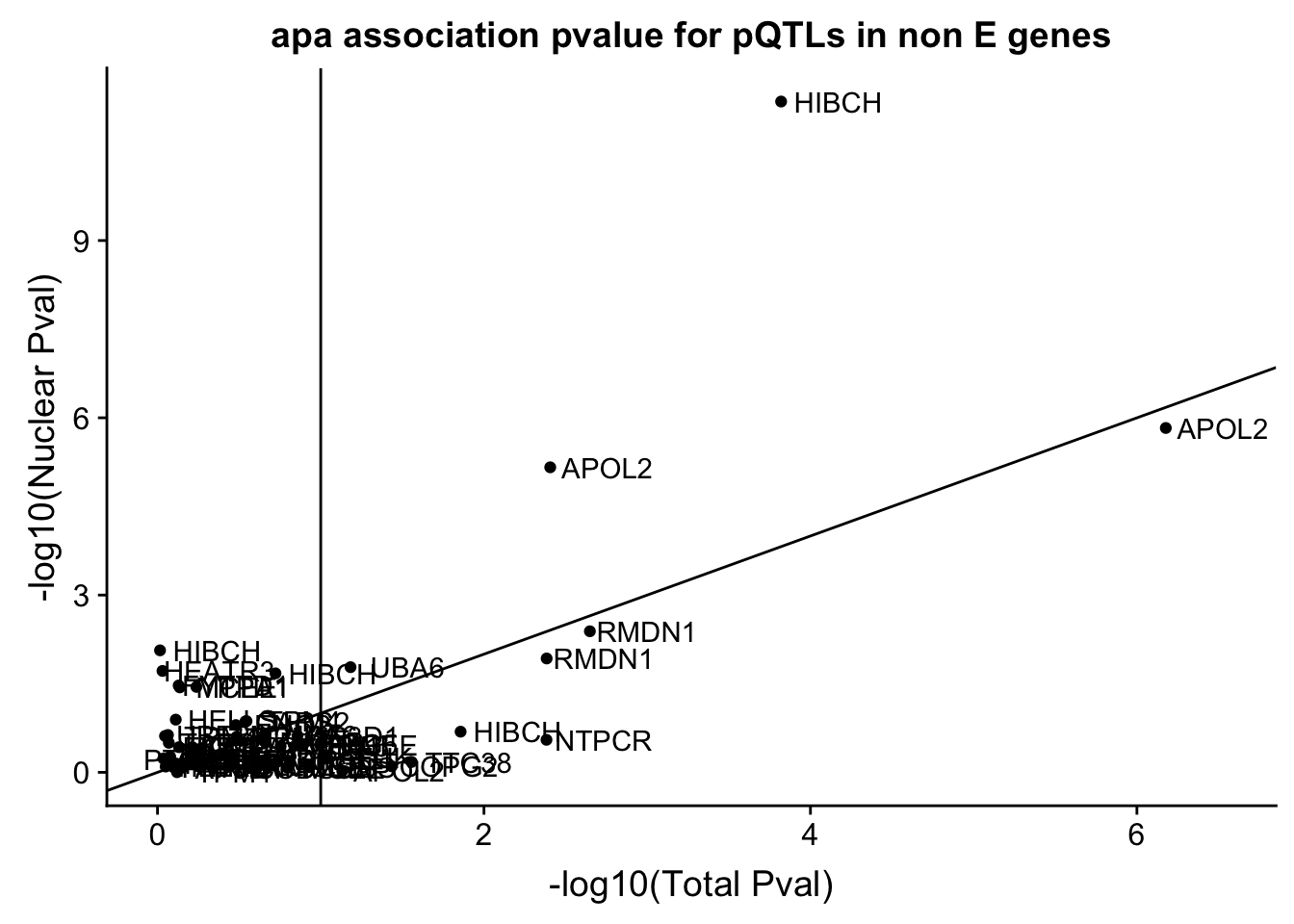
ggplot(pQTLBoth_notE,aes(x=TotalfixedPval,y=NuclearfixedPval)) + geom_point() + geom_abline(slope=1) + labs(x="-log10(Total Pval)",y="-log10(Nuclear Pval)", title="apa association pvalue for pQTLs in non E genes") + geom_vline(xintercept =1)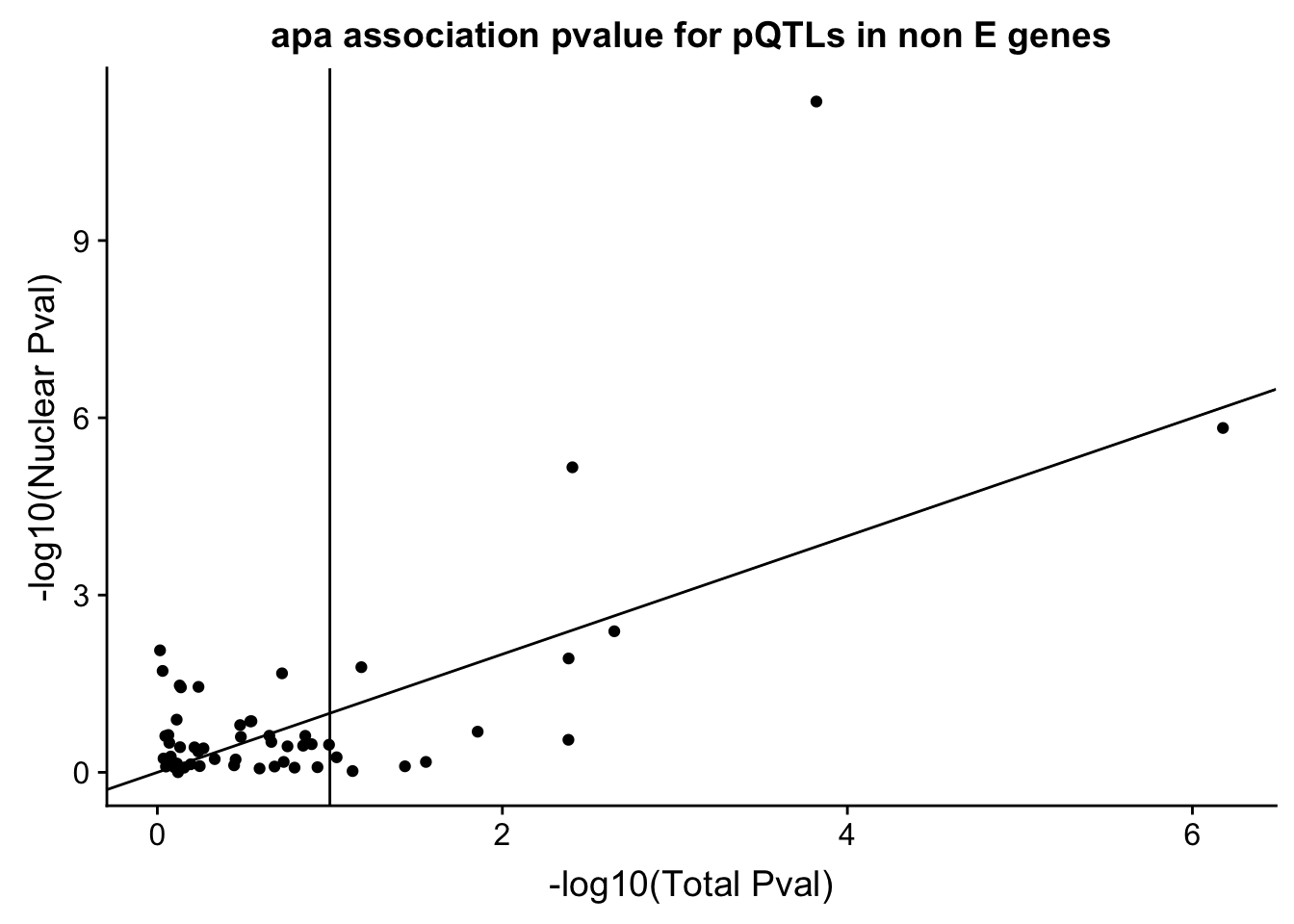
#+ geom_text(aes(label=gene),nudge_x = .35)
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.14.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] cowplot_0.9.4 reshape2_1.4.3 forcats_0.4.0 stringr_1.4.0
[5] dplyr_0.8.0.1 purrr_0.3.1 readr_1.3.1 tidyr_0.8.3
[9] tibble_2.0.1 ggplot2_3.1.0 tidyverse_1.2.1 workflowr_1.2.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.0 cellranger_1.1.0 plyr_1.8.4 pillar_1.3.1
[5] compiler_3.5.1 git2r_0.24.0 tools_3.5.1 digest_0.6.18
[9] lubridate_1.7.4 jsonlite_1.6 evaluate_0.13 nlme_3.1-137
[13] gtable_0.2.0 lattice_0.20-38 pkgconfig_2.0.2 rlang_0.3.1
[17] cli_1.0.1 rstudioapi_0.9.0 yaml_2.2.0 haven_2.1.0
[21] xfun_0.5 withr_2.1.2 xml2_1.2.0 httr_1.4.0
[25] knitr_1.21 hms_0.4.2 generics_0.0.2 fs_1.2.6
[29] rprojroot_1.3-2 grid_3.5.1 tidyselect_0.2.5 glue_1.3.0
[33] R6_2.4.0 readxl_1.3.0 rmarkdown_1.11 modelr_0.1.4
[37] magrittr_1.5 whisker_0.3-2 backports_1.1.3 scales_1.0.0
[41] htmltools_0.3.6 rvest_0.3.2 assertthat_0.2.0 colorspace_1.4-0
[45] labeling_0.3 stringi_1.3.1 lazyeval_0.2.1 munsell_0.5.0
[49] broom_0.5.1 crayon_1.3.4