Total vs Nuclear Example plots
Briana Mittleman
4/3/2019
Last updated: 2019-04-08
Checks: 6 0
Knit directory: threeprimeseq/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.2.0). The Report tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(12345) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/figure/
Ignored: data/.DS_Store
Ignored: data/perm_QTL_trans_noMP_5percov/
Ignored: output/.DS_Store
Untracked files:
Untracked: KalistoAbundance18486.txt
Untracked: analysis/4suDataIGV.Rmd
Untracked: analysis/AdaptTonyExampleplots.Rmd
Untracked: analysis/DirectionapaQTL.Rmd
Untracked: analysis/EvaleQTLs.Rmd
Untracked: analysis/YL_QTL_test.Rmd
Untracked: analysis/groSeqAnalysis.Rmd
Untracked: analysis/ncbiRefSeq_sm.sort.mRNA.bed
Untracked: analysis/peaksWithMisprimming.Rmd
Untracked: analysis/snake.config.notes.Rmd
Untracked: analysis/verifyBAM.Rmd
Untracked: analysis/verifybam_dubs.Rmd
Untracked: code/PeaksToCoverPerReads.py
Untracked: code/strober_pc_pve_heatmap_func.R
Untracked: data/18486.genecov.txt
Untracked: data/APApeaksYL.total.inbrain.bed
Untracked: data/AllPeak_counts/
Untracked: data/ApaQTLs/
Untracked: data/ApaQTLs_otherPhen/
Untracked: data/CTCF/
Untracked: data/ChromHmmOverlap/
Untracked: data/DistTXN2Peak_genelocAnno/
Untracked: data/EmpiricalDists/
Untracked: data/ExampleQTLplot2/
Untracked: data/FeatureoverlapPeaks/
Untracked: data/GM12878.chromHMM.bed
Untracked: data/GM12878.chromHMM.txt
Untracked: data/GWAS_overlap/
Untracked: data/LianoglouLCL/
Untracked: data/LocusZoom/
Untracked: data/LocusZoom_Unexp/
Untracked: data/LocusZoom_proc/
Untracked: data/MatchedSnps/
Untracked: data/NucSpecQTL/
Untracked: data/NuclearApaQTLs.txt
Untracked: data/PeakCounts/
Untracked: data/PeakCounts_noMP_5perc/
Untracked: data/PeakCounts_noMP_genelocanno/
Untracked: data/PeakUsage/
Untracked: data/PeakUsage_noMP/
Untracked: data/PeakUsage_noMP_GeneLocAnno/
Untracked: data/PeaksUsed/
Untracked: data/PeaksUsed_noMP_5percCov/
Untracked: data/PolyA_DB/
Untracked: data/QTL_overlap/
Untracked: data/RNAdecay/
Untracked: data/RNAkalisto/
Untracked: data/RefSeq_annotations/
Untracked: data/Replicates_usage/
Untracked: data/Signal_Loc/
Untracked: data/TNcompExamp/
Untracked: data/TotalApaQTLs.txt
Untracked: data/Totalpeaks_filtered_clean.bed
Untracked: data/UnderstandPeaksQC/
Untracked: data/WASP_STAT/
Untracked: data/YL-SP-18486-T-combined-genecov.txt
Untracked: data/YL-SP-18486-T_S9_R1_001-genecov.txt
Untracked: data/YL_QTL_test/
Untracked: data/apaExamp/
Untracked: data/apaExamp_proc/
Untracked: data/apaQTL_examp_noMP/
Untracked: data/bedgraph_peaks/
Untracked: data/bin200.5.T.nuccov.bed
Untracked: data/bin200.Anuccov.bed
Untracked: data/bin200.nuccov.bed
Untracked: data/clean_peaks/
Untracked: data/comb_map_stats.csv
Untracked: data/comb_map_stats.xlsx
Untracked: data/comb_map_stats_39ind.csv
Untracked: data/combined_reads_mapped_three_prime_seq.csv
Untracked: data/diff_iso_GeneLocAnno/
Untracked: data/diff_iso_proc/
Untracked: data/diff_iso_trans/
Untracked: data/eQTL_inAPA/
Untracked: data/eQTLs_Lietal/
Untracked: data/ensemble_to_genename.txt
Untracked: data/example_gene_peakQuant/
Untracked: data/explainProtVar/
Untracked: data/filtPeakOppstrand_cov_noMP_GeneLocAnno_5perc/
Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.bed
Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.noties.bed
Untracked: data/first50lines_closest.txt
Untracked: data/gencov.test.csv
Untracked: data/gencov.test.txt
Untracked: data/gencov_zero.test.csv
Untracked: data/gencov_zero.test.txt
Untracked: data/gene_cov/
Untracked: data/joined
Untracked: data/leafcutter/
Untracked: data/merged_combined_YL-SP-threeprimeseq.bg
Untracked: data/molPheno_noMP/
Untracked: data/mol_overlap/
Untracked: data/mol_pheno/
Untracked: data/nom_QTL/
Untracked: data/nom_QTL_opp/
Untracked: data/nom_QTL_trans/
Untracked: data/nuc6up/
Untracked: data/nuc_10up/
Untracked: data/other_qtls/
Untracked: data/pQTL_inAPA/
Untracked: data/pQTL_otherphen/
Untracked: data/pacbio_cov/
Untracked: data/peakPerRefSeqGene/
Untracked: data/peaks4DT/
Untracked: data/perm_QTL/
Untracked: data/perm_QTL_GeneLocAnno_noMP_5percov/
Untracked: data/perm_QTL_GeneLocAnno_noMP_5percov_3UTR/
Untracked: data/perm_QTL_diffWindow/
Untracked: data/perm_QTL_opp/
Untracked: data/perm_QTL_trans/
Untracked: data/perm_QTL_trans_filt/
Untracked: data/protAndAPAAndExplmRes.Rda
Untracked: data/protAndAPAlmRes.Rda
Untracked: data/protAndExpressionlmRes.Rda
Untracked: data/reads_mapped_three_prime_seq.csv
Untracked: data/smash.cov.results.bed
Untracked: data/smash.cov.results.csv
Untracked: data/smash.cov.results.txt
Untracked: data/smash_testregion/
Untracked: data/ssFC200.cov.bed
Untracked: data/temp.file1
Untracked: data/temp.file2
Untracked: data/temp.gencov.test.txt
Untracked: data/temp.gencov_zero.test.txt
Untracked: data/threePrimeSeqMetaData.csv
Untracked: data/threePrimeSeqMetaData55Ind.txt
Untracked: data/threePrimeSeqMetaData55Ind.xlsx
Untracked: data/threePrimeSeqMetaData55Ind_noDup.txt
Untracked: data/threePrimeSeqMetaData55Ind_noDup.xlsx
Untracked: data/threePrimeSeqMetaData55Ind_noDup_WASPMAP.txt
Untracked: data/threePrimeSeqMetaData55Ind_noDup_WASPMAP.xlsx
Untracked: data/threePrimeSeqMetaData55Ind_redobatch4.xlsx
Untracked: data/~$threePrimeSeqMetaData55Ind_redobatch4.xlsx
Untracked: manuscript/
Untracked: output/APAqtlExamp/
Untracked: output/LZ/
Untracked: output/deeptools_plots/
Untracked: output/picard/
Untracked: output/plots/
Untracked: output/qual.fig2.pdf
Unstaged changes:
Modified: analysis/28ind.peak.explore.Rmd
Modified: analysis/CompareLianoglouData.Rmd
Modified: analysis/ExampleQTLPlot2.Rmd
Modified: analysis/HistoneModandPAS.Rmd
Modified: analysis/NewPeakPostMP.Rmd
Modified: analysis/NuclearSpecQTL.Rmd
Modified: analysis/PeakToXper.Rmd
Modified: analysis/RNAdecayAndAPA.Rmd
Modified: analysis/apaQTLoverlapGWAS.Rmd
Modified: analysis/characterize_apaQTLs.Rmd
Modified: analysis/cleanupdtseq.internalpriming.Rmd
Modified: analysis/coloc_apaQTLs_protQTLs.Rmd
Modified: analysis/dif.iso.usage.leafcutter.Rmd
Modified: analysis/diffIsoAnalysisNewMapping.Rmd
Modified: analysis/diff_iso_pipeline.Rmd
Modified: analysis/explainpQTLs.Rmd
Modified: analysis/explore.filters.Rmd
Modified: analysis/fixBWChromNames.Rmd
Modified: analysis/flash2mash.Rmd
Modified: analysis/initialPacBioQuant.Rmd
Modified: analysis/mispriming_approach.Rmd
Modified: analysis/overlapMolQTL.Rmd
Modified: analysis/overlapMolQTL.opposite.Rmd
Modified: analysis/overlap_qtls.Rmd
Modified: analysis/peakOverlap_oppstrand.Rmd
Modified: analysis/peakQCPPlots.Rmd
Modified: analysis/pheno.leaf.comb.Rmd
Modified: analysis/pipeline_55Ind.Rmd
Modified: analysis/swarmPlots_QTLs.Rmd
Modified: analysis/test.max2.Rmd
Modified: analysis/test.smash.Rmd
Modified: analysis/understandPeaks.Rmd
Modified: analysis/unexplainedeQTL_analysis.Rmd
Modified: code/Snakefile
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 4568d1f | Briana Mittleman | 2019-04-08 | add boxplot and genometrack |
| html | 0ff28d4 | Briana Mittleman | 2019-04-03 | Build site. |
| Rmd | a79791b | Briana Mittleman | 2019-04-03 | start tot nuc example plots |
library(tidyverse)── Attaching packages ──────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.0 ✔ purrr 0.3.1
✔ tibble 2.0.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.4.0
✔ readr 1.3.1 ✔ forcats 0.4.0 Warning: package 'tibble' was built under R version 3.5.2Warning: package 'tidyr' was built under R version 3.5.2Warning: package 'purrr' was built under R version 3.5.2Warning: package 'dplyr' was built under R version 3.5.2Warning: package 'stringr' was built under R version 3.5.2Warning: package 'forcats' was built under R version 3.5.2── Conflicts ─────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(reshape2)
Attaching package: 'reshape2'The following object is masked from 'package:tidyr':
smithsHeatmap in one gene
I want to make an example heatmap for the total vs nuclear difference similar to the ones I did for the qtls.
I will take a similar approach where I make one then create a script to make it for all examples
chr21:43762910:43762982:TFF2
Count data is in:
/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_processed_GeneLocAnno_bothFrac/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_sm_quant_processed_fixed.fc
grep TFF2 /project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_processed_GeneLocAnno_bothFrac/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_sm_quant_processed_fixed.fc > /project2/gilad/briana/threeprimeseq/data/TNcompExamp/TNcomp_TFF2.txtI will need to divide by the mapped read counts for the library:
metadata=read.table("../data/threePrimeSeqMetaData55Ind_noDup_WASPMAP.txt",header = T) %>% select(Sample_ID,Mapped_noMP )
metadata_melt=melt(metadata, id.vars = c("Sample_ID"), value.name = "MappedRead") %>% mutate(MappPM=MappedRead/10^6)Header file:
TNhead=read.table("../data/TNcompExamp/TNCountheader.txt", header = T,stringsAsFactors = F)read in data and melt it:
TN_TFF2=read.table("../data/TNcompExamp/TNcomp_TFF2.txt", col.names =colnames(TNhead),stringsAsFactors = F) %>% select(-Chr,-Geneid,-Strand, -Length)
TN_TFF2$Start=as.character(TN_TFF2$Start)
TN_TFF2$End=as.character(TN_TFF2$End)
TN_TFF2= TN_TFF2 %>% mutate(PeakLoc= paste(Start,End,sep="_")) %>% select(-Start, -End)
TN_TFF2_melt=melt(TN_TFF2, id.vars =c("PeakLoc"), variable.name = "ID", value.name = "PeakCount" ) %>% mutate(Sample_ID=substr(ID, 2, length(ID))) Join:
TN_TFF2_withMeta=TN_TFF2_melt %>% inner_join(metadata_melt, by=c("Sample_ID")) %>% mutate(Fraction=ifelse(grepl("T",Sample_ID), "Total","Nuclear")) %>% mutate(NormCount=PeakCount/MappPM) %>% group_by(PeakLoc,Fraction) %>% summarise(meanCPM=mean(NormCount))Warning: Column `Sample_ID` joining character vector and factor, coercing
into character vectormy_palette <- colorRampPalette(c("white", "khaki1", "orange", "red", "darkred", "black"))
ggplot(TN_TFF2_withMeta, aes(x=PeakLoc, y=Fraction)) + geom_tile(aes(fill = meanCPM))+ scale_fill_gradientn(colors =my_palette(100)) + theme(axis.text.x = element_text(angle = 90, hjust = 1)) + labs(x="PAS", title="TFF2")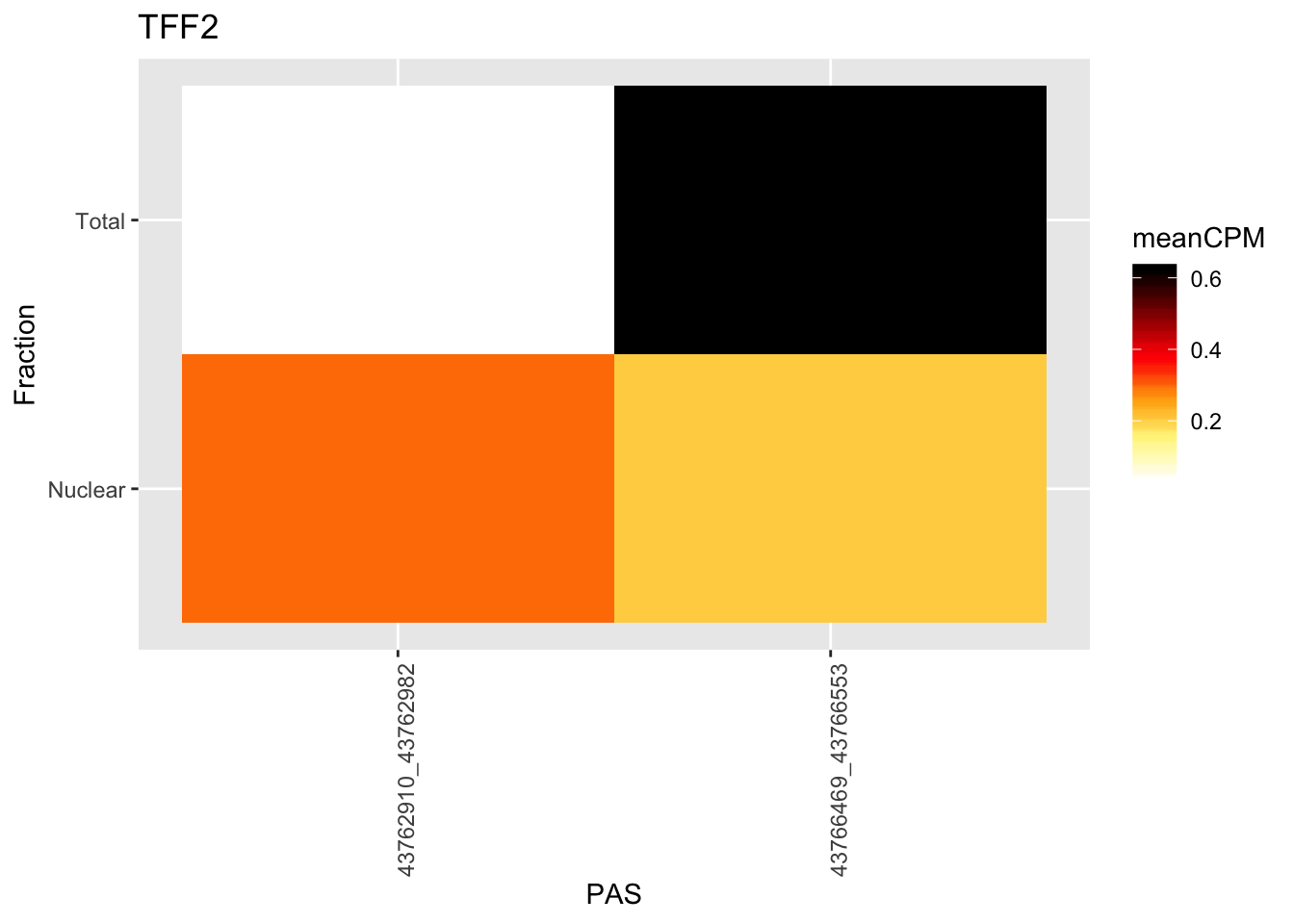
| Version | Author | Date |
|---|---|---|
| 0ff28d4 | Briana Mittleman | 2019-04-03 |
Super low expression of this gene. Better example when it is
Generalize:
TvNHeatmap.R
library(tidyverse)
library(reshape2)
library(optparse)
library(cowplot)
option_list = list(
make_option(c("-P", "--pheno"), action="store", default=NA, type='character',
help="input pheno file"),
make_option(c("-g", "--gene"), action="store", default=NA, type='character',
help="gene"),
make_option(c("-o", "--output"), action="store", default=NA, type='character',
help="output file for plot")
)
opt_parser <- OptionParser(option_list=option_list)
opt <- parse_args(opt_parser)
metadata=read.table("/project2/gilad/briana/threeprimeseq/data/TNcompExamp/threePrimeSeqMetaData55Ind_noDup_WASPMAP.txt",header=T, stringsAsFactors = F) %>% select(Sample_ID,Mapped_noMP )
metadata_melt=melt(metadata, id.vars = c("Sample_ID"), value.name = "MappedRead") %>% mutate(MappPM=MappedRead/10^6)
TNhead=read.table("/project2/gilad/briana/threeprimeseq/data/TNcompExamp/TNCountheader.txt", header = T,stringsAsFactors = F)
TN=read.table(opt$pheno, col.names =colnames(TNhead),stringsAsFactors = F) %>% select(-Chr,-Geneid,-Strand, -Length)
TN$Start=as.character(TN$Start)
TN$End=as.character(TN$End)
TN= TN%>% mutate(PeakLoc= paste(Start,End,sep="-")) %>% select(-Start, -End)
TN_melt=melt(TN, id.vars =c("PeakLoc"), variable.name = "ID", value.name = "PeakCount" ) %>% mutate(Sample_ID=substr(ID, 2, length(ID)))
TN_withMeta=TN_melt %>% inner_join(metadata_melt, by=c("Sample_ID")) %>% mutate(Fraction=ifelse(grepl("T",Sample_ID), "Total","Nuclear")) %>% mutate(NormCount=PeakCount/MappPM) %>% group_by(PeakLoc,Fraction) %>% summarise(meanCPM=mean(NormCount))
my_palette <- colorRampPalette(c("white", "khaki1", "orange", "red", "darkred", "black"))
outplot=ggplot(TN_withMeta, aes(x=PeakLoc, y=Fraction)) + geom_tile(aes(fill = meanCPM))+ scale_fill_gradientn(colors =my_palette(100)) + theme(axis.text.x = element_text(angle = 90, hjust = 1)) + labs(x="PAS", title=opt$gene)
ggsave(plot=outplot, filename=opt$output, height=10, width=10)Script to make phenotype file and run R script:
TvNMakeHeatmap.sh
#!/bin/bash
#SBATCH --job-name=TvNMakeHeatmap
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=TvNMakeHeatmap.out
#SBATCH --error=TvNMakeHeatmap.err
#SBATCH --partition=broadwl
#SBATCH --mem=18G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
gene=$1
grep ${gene} /project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_processed_GeneLocAnno_bothFrac/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_sm_quant_processed_fixed.fc > /project2/gilad/briana/threeprimeseq/data/TNcompExamp/TNcomp_${gene}.txt
Rscript TvNHeatmap.R -P /project2/gilad/briana/threeprimeseq/data/TNcompExamp/TNcomp_${gene}.txt -g ${gene} -o /project2/gilad/briana/threeprimeseq/data/TNcompExamp/TNcomp_${gene}_heatmap.png
Examples to test
diffIso=read.table(file="../data/diff_iso_GeneLocAnno/SigPeaksHigherInNuc.txt", header = T) %>% arrange(deltapsi)head(diffIso) intron logef Nuclear Total
1 chr1:151134497:151134579:TNFAIP8L2 -1.531127 0.7805416 0.1426529
2 chr21:43762910:43762982:TFF2 -1.292723 0.7517177 0.1857824
3 chr3:23306502:23306675:UBE2E2 -1.576854 0.6895186 0.1527728
4 chr14:67029307:67029417:GPHN -1.178720 0.7952505 0.2688294
5 chr6:84007319:84007404:ME1 -1.941535 0.6378959 0.1158490
6 chr7:73885912:73885994:GTF2IRD1 -1.094156 0.8030045 0.3136450
deltapsi
1 -0.6378887
2 -0.5659353
3 -0.5367458
4 -0.5264211
5 -0.5220469
6 -0.4893595sbatch TvNMakeHeatmap.sh GPHN
sbatch TvNMakeHeatmap.sh ME1
sbatch TvNMakeHeatmap.sh GTF2IRD1
sbatch TvNMakeHeatmap.sh DOCK9
sbatch TvNMakeHeatmap.sh UNQ6494Track image with pygenome
I want to make normalized BW to plot on tracks. This will be easier to show for this. I can use bamCoverage in deeptools and normalize to cpm.
normbam2BW.sh
#!/bin/bash
#SBATCH --job-name=normbam2BW
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=normbam2BW.out
#SBATCH --error=normbam2BW.err
#SBATCH --partition=broadwl
#SBATCH --mem=18G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
i=$1
bamCoverage -b /project2/gilad/briana/threeprimeseq/data/sort/YL-SP-$i-sort.bam -o /project2/gilad/briana/threeprimeseq/data/normalizedBW/YL-SP-$i-norm.bw \
--normalizeUsingRPKMRun on all file is /project2/gilad/briana/threeprimeseq/data/sort/
(need to move these)
run_normbam2BW.sh
#!/bin/bash
#SBATCH --job-name=run_normbam2BW
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=run_normbam2BW.out
#SBATCH --error=run_normbam2BW.err
#SBATCH --partition=broadwl
#SBATCH --mem=10G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
for i in $(ls /project2/gilad/briana/threeprimeseq/data/sort/*.bam)
do
describer=$(echo ${i} | sed -e 's/.*YL-SP-//' | sed -e "s/-sort.bam//")
sbatch normbam2BW.sh $describer
doneMake a merged version.
/project2/gilad/briana/threeprimeseq/data/normalizedBW_merged
mergeNormBW.sh
#!/bin/bash
#SBATCH --job-name=mergeNormBW
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=mergeNormBW.out
#SBATCH --error=mergeNormBW.err
#SBATCH --partition=broadwl
#SBATCH --mem=10G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
ls -d -1 /project2/gilad/briana/threeprimeseq/data/normalizedBW/*T* | tail -n +2 > /project2/gilad/briana/threeprimeseq/data/list_bw/list_of_TotNorombigwig.txt
ls -d -1 /project2/gilad/briana/threeprimeseq/data/normalizedBW/*N* | tail -n +2 > /project2/gilad/briana/threeprimeseq/data/list_bw/list_of_NucNorombigwig.txt
bigWigMerge -inList /project2/gilad/briana/threeprimeseq/data/list_bw/list_of_TotNorombigwig.txt /project2/gilad/briana/threeprimeseq/data/normalizedBW_merged/Total_NormalizedMerged.bg
sort -k1,1 -k2,2n /project2/gilad/briana/threeprimeseq/data/normalizedBW_merged/Total_NormalizedMerged.bg > /project2/gilad/briana/threeprimeseq/data/normalizedBW_merged/Total_NormalizedMerged.sort.bg
bedGraphToBigWig /project2/gilad/briana/threeprimeseq/data/normalizedBW_merged/Total_NormalizedMerged.sort.bg /project2/gilad/briana/genome_anotation_data/chrom.length.txt /project2/gilad/briana/threeprimeseq/data/normalizedBW_merged/Total_NormalizedMerged.bw
bigWigMerge -inList /project2/gilad/briana/threeprimeseq/data/list_bw/list_of_NucNorombigwig.txt /project2/gilad/briana/threeprimeseq/data/normalizedBW_merged/Nuclear_NormalizedMerged.bg
sort -k1,1 -k2,2n /project2/gilad/briana/threeprimeseq/data/normalizedBW_merged/Nuclear_NormalizedMerged.bg > /project2/gilad/briana/threeprimeseq/data/normalizedBW_merged/Nuclear_NormalizedMerged.sort.bg
bedGraphToBigWig /project2/gilad/briana/threeprimeseq/data/normalizedBW_merged/Nuclear_NormalizedMerged.sort.bg /project2/gilad/briana/genome_anotation_data/chrom.length.txt /project2/gilad/briana/threeprimeseq/data/normalizedBW_merged/Nuclear_NormalizedMerged.bwUse pygenome tracks:
makeTNcompINIfile.sh
#!/bin/bash
#SBATCH --job-name=makeTNcompINIfile
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=makeTNcompINIfile.out
#SBATCH --error=makeTNcompINIfile.err
#SBATCH --partition=broadwl
#SBATCH --mem=10G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
make_tracks_file --trackFiles /project2/gilad/briana/threeprimeseq/data/normalizedBW_merged/Nuclear_NormalizedMerged.bw /project2/gilad/briana/threeprimeseq/data/normalizedBW_merged/Total_NormalizedMerged.bw /project2/gilad/briana/threeprimeseq/data/normalizedBW/YL-SP-18499-N-combined-norm.bw /project2/gilad/briana/threeprimeseq/data/normalizedBW/YL-SP-18499-T-combined-norm.bw /project2/gilad/briana/threeprimeseq/data/normalizedBW/YL-SP-19128-N-combined-norm.bw /project2/gilad/briana/threeprimeseq/data/normalizedBW/YL-SP-19128-T-combined-norm.bw /project2/gilad/briana/threeprimeseq/data/normalizedBW/YL-SP-18516-N-combined-norm.bw /project2/gilad/briana/threeprimeseq/data/normalizedBW/YL-SP-18516-T-combined-norm.bw /project2/gilad/briana/threeprimeseq/data/normalizedBW/YL-SP-18912-N-combined-norm.bw /project2/gilad/briana/threeprimeseq/data/normalizedBW/YL-SP-18912-T-combined-norm.bw /project2/gilad/briana/genome_anotation_data/NCBI_refseq_forPyGenTrack_sort.bed -o /project2/gilad/briana/threeprimeseq/data/TNcompExamp/TotalvNuc_exmple.ini
#/project2/gilad/briana/threeprimeseq/data/peaks4DT/APAPeaks_5percCov_fixedStrand.bed Can add
/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Total_fixed.pheno.5percPeaks.txt
/project2/gilad/briana/threeprimeseq/data/phenotypes_filtPeakTranscript_noMP_GeneLocAnno/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno.NoMP_sm_quant.Nuclear_fixed.pheno.5percPeaks.txt
Would need to convert to bed
try on a region:
DOCK9
get region from iGV for now
pyGenomeTracks --tracks TotalvNuc_exmple.ini --region 13:99,439,464-99,736,383 -out testDock9.png
pyGenomeTracks --tracks TotalvNuc_exmple.ini --region 14:66,968,548-67,658,985 -out testGPHN.png
pyGenomeTracks --tracks TotalvNuc_exmple.ini --region 21:43,766,162-43,772,074 -out testTFF2.png
pyGenomeTracks --tracks TotalvNuc_exmple.ini --region 9:92,252,698-92,336,674 -out testUNQ6495.png
chr14:66,968,548-67,658,985
Create boxplots
TN_TFF2_withMetaBox=TN_TFF2_melt %>% inner_join(metadata_melt, by=c("Sample_ID")) %>% mutate(Fraction=ifelse(grepl("T",Sample_ID), "Total","Nuclear"))Warning: Column `Sample_ID` joining character vector and factor, coercing
into character vectorggplot(TN_TFF2_withMetaBox, aes(x=Fraction, y=MappPM, fill=Fraction)) + geom_boxplot(width=.45) + geom_jitter() + scale_fill_brewer(palette = "YlOrRd") + labs(y="CPM", title="TFF2")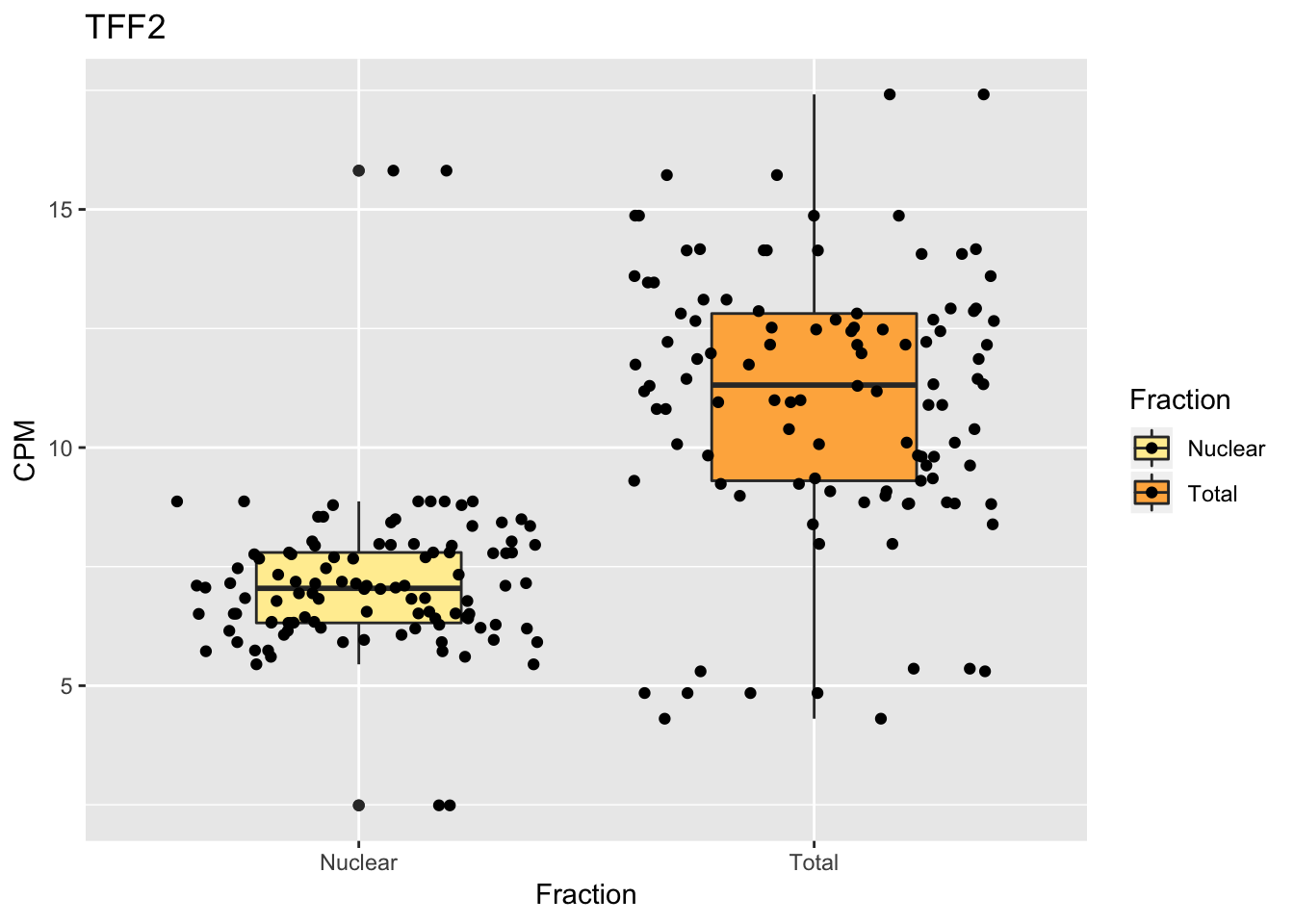
R script:
TvNBoxplot.R
library(tidyverse)
library(reshape2)
library(optparse)
library(cowplot)
option_list = list(
make_option(c("-P", "--pheno"), action="store", default=NA, type='character',
help="input pheno file"),
make_option(c("-g", "--gene"), action="store", default=NA, type='character',
help="gene"),
make_option(c("-o", "--output"), action="store", default=NA, type='character',
help="output file for plot")
)
opt_parser <- OptionParser(option_list=option_list)
opt <- parse_args(opt_parser)
metadata=read.table("/project2/gilad/briana/threeprimeseq/data/TNcompExamp/threePrimeSeqMetaData55Ind_noDup_WASPMAP.txt",header=T, stringsAsFactors = F) %>% select(Sample_ID,Mapped_noMP )
metadata_melt=melt(metadata, id.vars = c("Sample_ID"), value.name = "MappedRead") %>% mutate(MappPM=MappedRead/10^6)
TNhead=read.table("/project2/gilad/briana/threeprimeseq/data/TNcompExamp/TNCountheader.txt", header = T,stringsAsFactors = F)
TN=read.table(opt$pheno, col.names =colnames(TNhead),stringsAsFactors = F) %>% select(-Chr,-Geneid,-Strand, -Length)
TN$Start=as.character(TN$Start)
TN$End=as.character(TN$End)
TN= TN%>% mutate(PeakLoc= paste(Start,End,sep="-")) %>% select(-Start, -End)
TN_melt=melt(TN, id.vars =c("PeakLoc"), variable.name = "ID", value.name = "PeakCount" ) %>% mutate(Sample_ID=substr(ID, 2, length(ID)))
TN_withMetaBox=TN_melt %>% inner_join(metadata_melt, by=c("Sample_ID")) %>% inner_join(metadata_melt, by=c("Sample_ID")) %>% mutate(Fraction=ifelse(grepl("T",Sample_ID), "Total","Nuclear"))
outplot=ggplot(TN_TFF2_withMetaBox, aes(x=Fraction, y=MappPM, fill=Fraction)) + geom_boxplot(width=.45) + geom_jitter() + scale_fill_brewer(palette = "YlOrRd") + labs(y="CPM", title=opt$gene)
ggsave(plot=outplot, filename=opt$output, height=10, width=10)TvNMakeHeatmapandBoxplot.sh
#!/bin/bash
#SBATCH --job-name=TvNMakeHeatmapandBoxplot
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=TvNMakeHeatmapandBoxplot.out
#SBATCH --error=TvNMakeHeatmapandBoxplot.err
#SBATCH --partition=broadwl
#SBATCH --mem=18G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
gene=$1
grep ${gene} /project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_processed_GeneLocAnno_bothFrac/filtered_APApeaks_merged_allchrom_refseqGenes.GeneLocAnno_sm_quant_processed_fixed.fc > /project2/gilad/briana/threeprimeseq/data/TNcompExamp/TNcomp_${gene}.txt
Rscript TvNHeatmap.R -P /project2/gilad/briana/threeprimeseq/data/TNcompExamp/TNcomp_${gene}.txt -g ${gene} -o /project2/gilad/briana/threeprimeseq/data/TNcompExamp/TNcomp_${gene}_heatmap.png
Rscript TvNBoxplot.R -P /project2/gilad/briana/threeprimeseq/data/TNcompExamp/TNcomp_${gene}.txt -g ${gene} -o /project2/gilad/briana/threeprimeseq/data/TNcompExamp/TNcomp_${gene}_boxplot.png
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.14.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] reshape2_1.4.3 forcats_0.4.0 stringr_1.4.0 dplyr_0.8.0.1
[5] purrr_0.3.1 readr_1.3.1 tidyr_0.8.3 tibble_2.0.1
[9] ggplot2_3.1.0 tidyverse_1.2.1
loaded via a namespace (and not attached):
[1] Rcpp_1.0.0 RColorBrewer_1.1-2 cellranger_1.1.0
[4] plyr_1.8.4 pillar_1.3.1 compiler_3.5.1
[7] git2r_0.24.0 workflowr_1.2.0 tools_3.5.1
[10] digest_0.6.18 lubridate_1.7.4 jsonlite_1.6
[13] evaluate_0.13 nlme_3.1-137 gtable_0.2.0
[16] lattice_0.20-38 pkgconfig_2.0.2 rlang_0.3.1
[19] cli_1.0.1 rstudioapi_0.9.0 yaml_2.2.0
[22] haven_2.1.0 xfun_0.5 withr_2.1.2
[25] xml2_1.2.0 httr_1.4.0 knitr_1.21
[28] hms_0.4.2 generics_0.0.2 fs_1.2.6
[31] rprojroot_1.3-2 grid_3.5.1 tidyselect_0.2.5
[34] glue_1.3.0 R6_2.4.0 readxl_1.3.0
[37] rmarkdown_1.11 modelr_0.1.4 magrittr_1.5
[40] whisker_0.3-2 backports_1.1.3 scales_1.0.0
[43] htmltools_0.3.6 rvest_0.3.2 assertthat_0.2.0
[46] colorspace_1.4-0 labeling_0.3 stringi_1.3.1
[49] lazyeval_0.2.1 munsell_0.5.0 broom_0.5.1
[52] crayon_1.3.4